
Chemical Reactions of Haloalkanes and Haloarenes
Quick Notes
- Haloalkanes undergo nucleophilic substitution, elimination, and metal-coupling reactions.
- Haloarenes react differently due to resonance and hybridisation effects.
- Reaction conditions (solvent, base strength, temperature) strongly influence the product pathway.
- Understanding the mechanisms (SN1, SN2, E2) is crucial for predicting reactivity.
Full Notes
Haloalkanes typically undergo three main types of reactions: nucleophilic substitution, elimination, and reactions with metals.
Nucleophilic Substitution Reactions
In these reactions, the halogen atom (which is more electronegative) is replaced by a nucleophile. This is one of the most fundamental types of reactions shown by haloalkanes.
General Reaction:
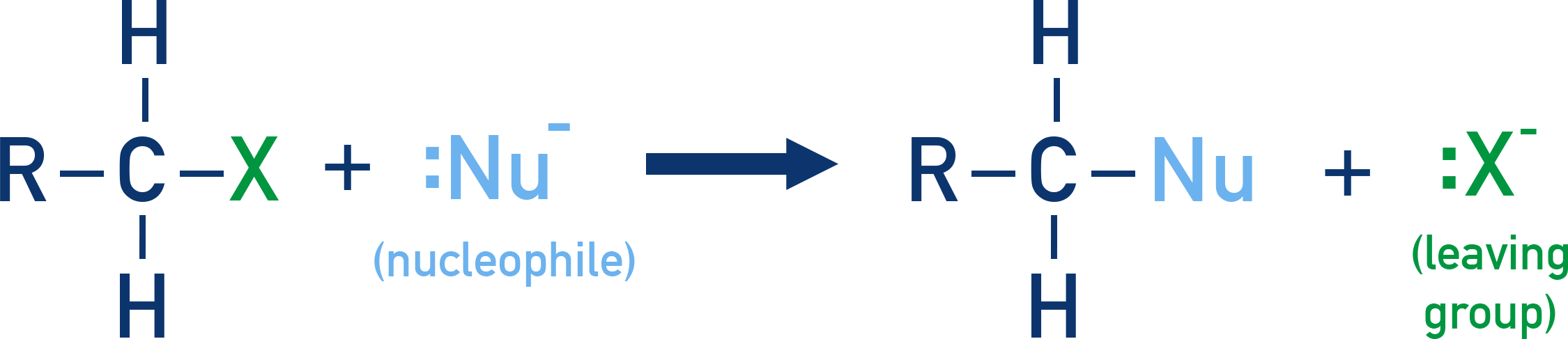
Examples of nucleophiles include OH−, CN−, NH3 and alkoxides
SN2 – Substitution Nucleophilic Bimolecular
The SN2 mechanism occurs in a single concerted step, where bond breaking and bond formation happen simultaneously.
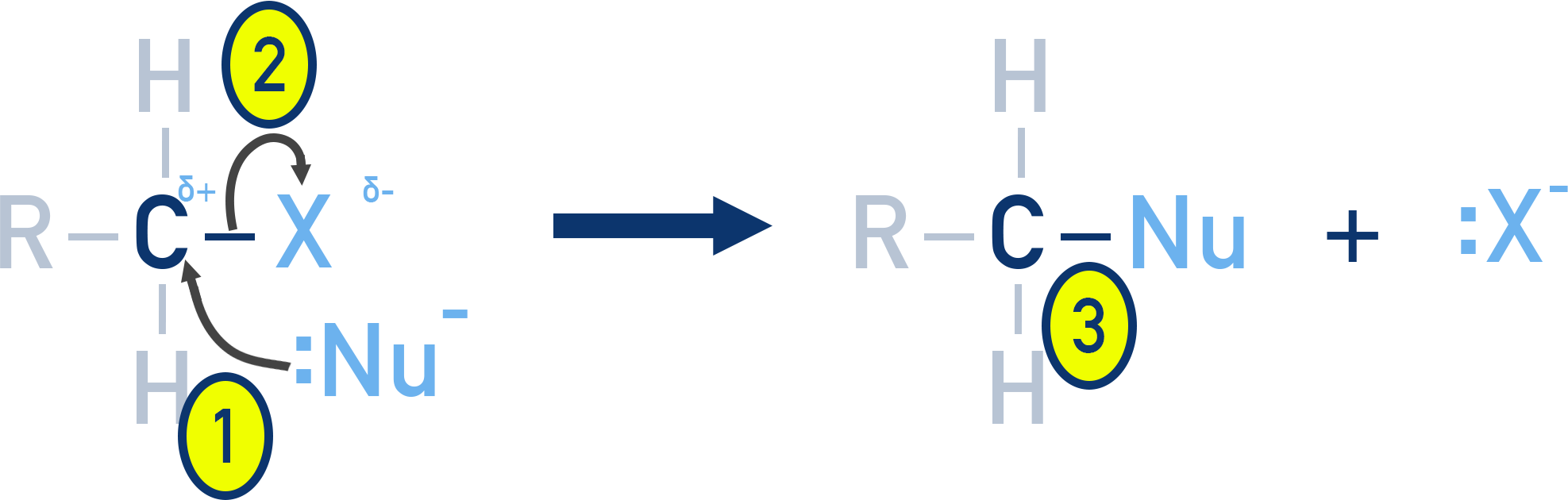
Occurs in one step (both reactants are involved in the same step).
- Curly arrow from nucleophile to δ+ carbon.
- Curly arrow from C–X bond to halogen (X− leaves).
- New bond forms between nucleophile and carbon.
The nucleophile attacks the carbon at the same time as the leaving group (halide) departs. The reaction results in inversion of configuration (Walden inversion).
A transition state is formed with partial bonds – both the nucleophile and the leaving group are briefly attached.
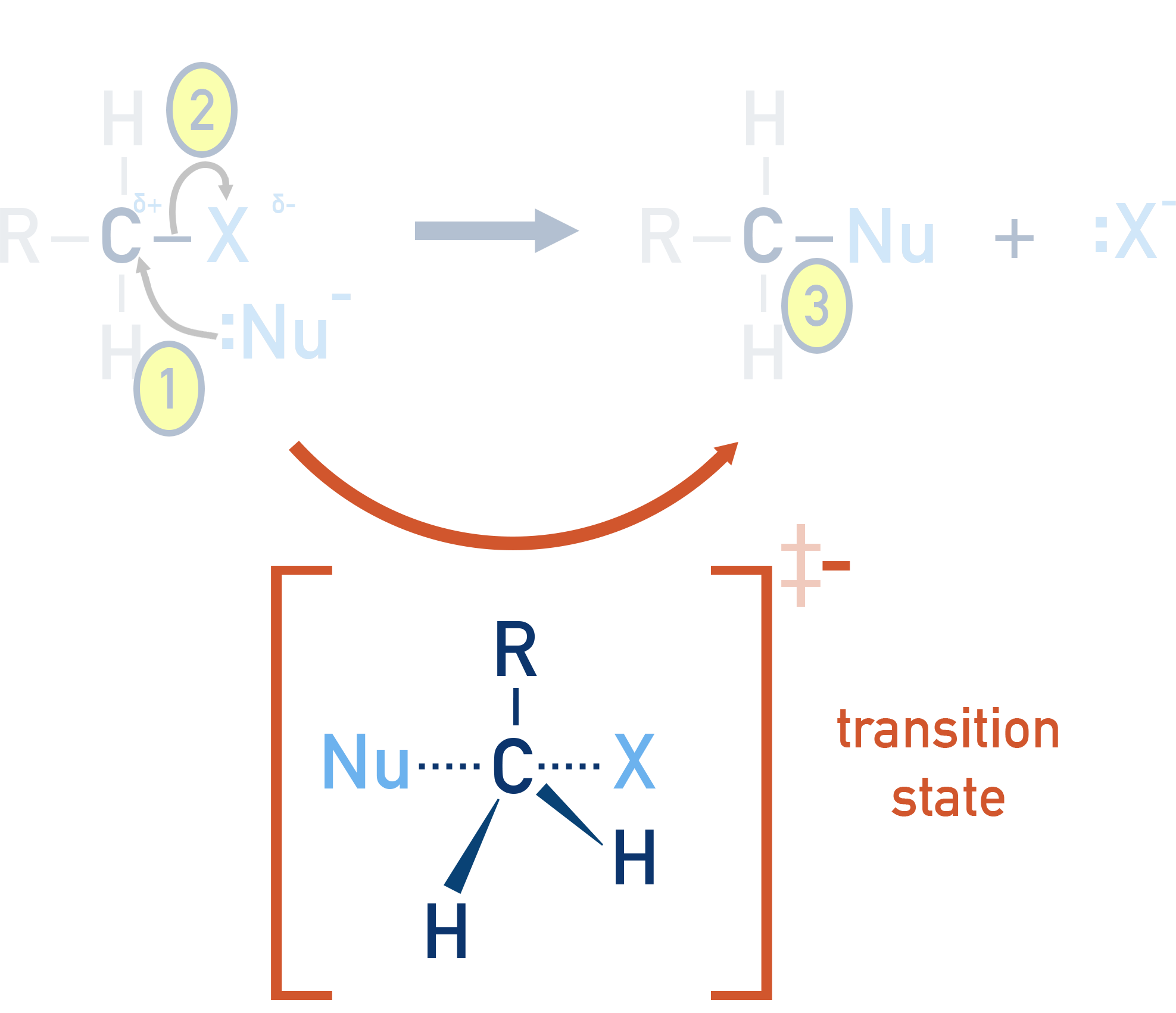
- Favoured by primary halogenoalkanes, where the central carbon is less hindered (‘blocked’) by other carbon atoms.
Example Formation of ethanol from bromoethane
CH3CH2Br + OH− → CH3CH2OH + Br−
Key point: Steric hindrance is low in primary halogenoalkanes, allowing the nucleophile to attack easily from the back. As a result, it most favoured for primary haloalkanes, where steric hindrance is minimal.
Rate Law: Rate = k[RX][Nu−]
SN1 – Substitution Nucleophilic Unimolecular
SN1 reactions proceed via a two-step mechanism and are common for tertiary haloalkanes where a stable carbocation can form.
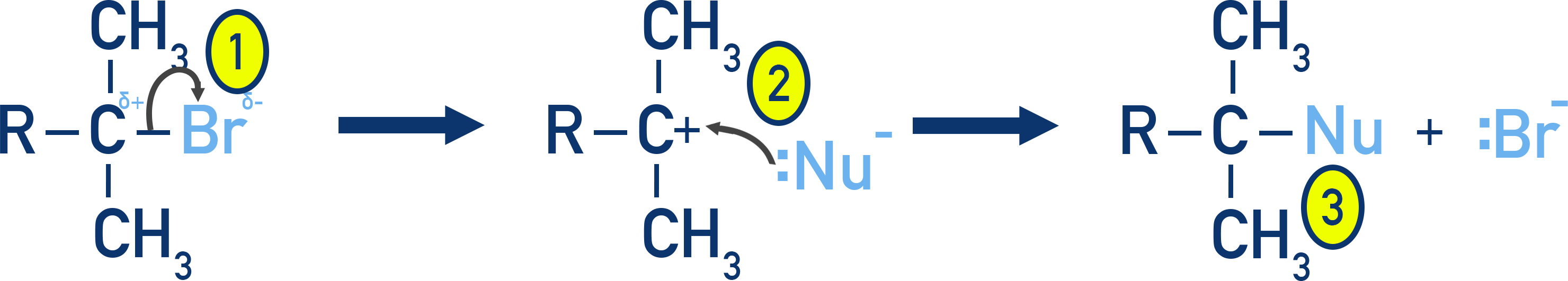
Occurs in two steps.
- The halide leaves first, forming a carbocation (slow step – rate-determining).
- The nucleophile then attacks the positively charged carbon (carbocation intermediate).
- New bond formed between the C and Nu.
Favoured by tertiary halogenoalkanes, where the carbocation is stabilised by alkyl groups via the positive inductive effect (electron-donating effect of surrounding methyl groups).
The carbocation intermediate is planar, so attack can happen from either side, forming a racemic mixture (loss of optical activity).
Rate Law: Rate = k[RX]

Primary halogenoalkanes can’t follow SN1 mechanisms as the positive inductive effect isn’t strong enough to stabilise a carbocation intermediate long enough for it to form and react with a nucleophile, meaning it has to react by SN2. Tertiary halogenoalkanes can’t follow SN2 mechanisms because the bulky carbon groups bonded to the C in the C-X group ‘block’ the incoming nucleophile (this is called steric hinderance).
Stereoisomers and Reaction Mechanisms: SN1 vs SN2
SN1 Reactions:
Proceed via a carbocation intermediate, which is planar. Nucleophilic attack can occur from either side with equal probability, forming the two possible stereoisomers (specifically optical isomers - see optical isomers and chiral centre’s for more detail) in equal amounts. This gives a racemic mixture.
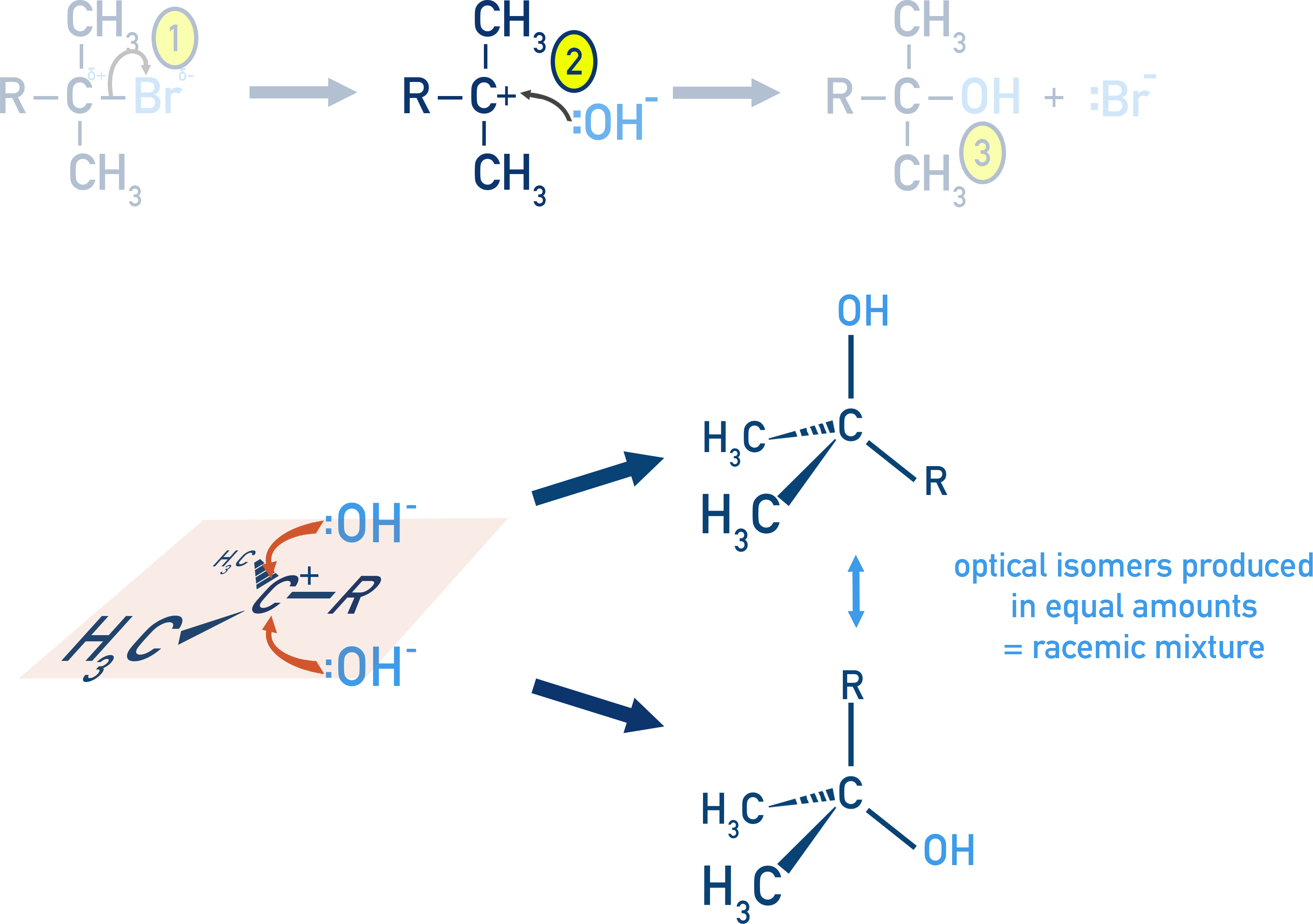
If a product mixture is optically inactive but formed from an optically active compound, it suggests an SN1 mechanism.
SN2 Reactions:
Involve a single-step mechanism where the nucleophile attacks from the opposite side to the leaving group. The incoming nucleophile ends up bonding in the opposite position to the leaving group, causing an inversion of the configuration.
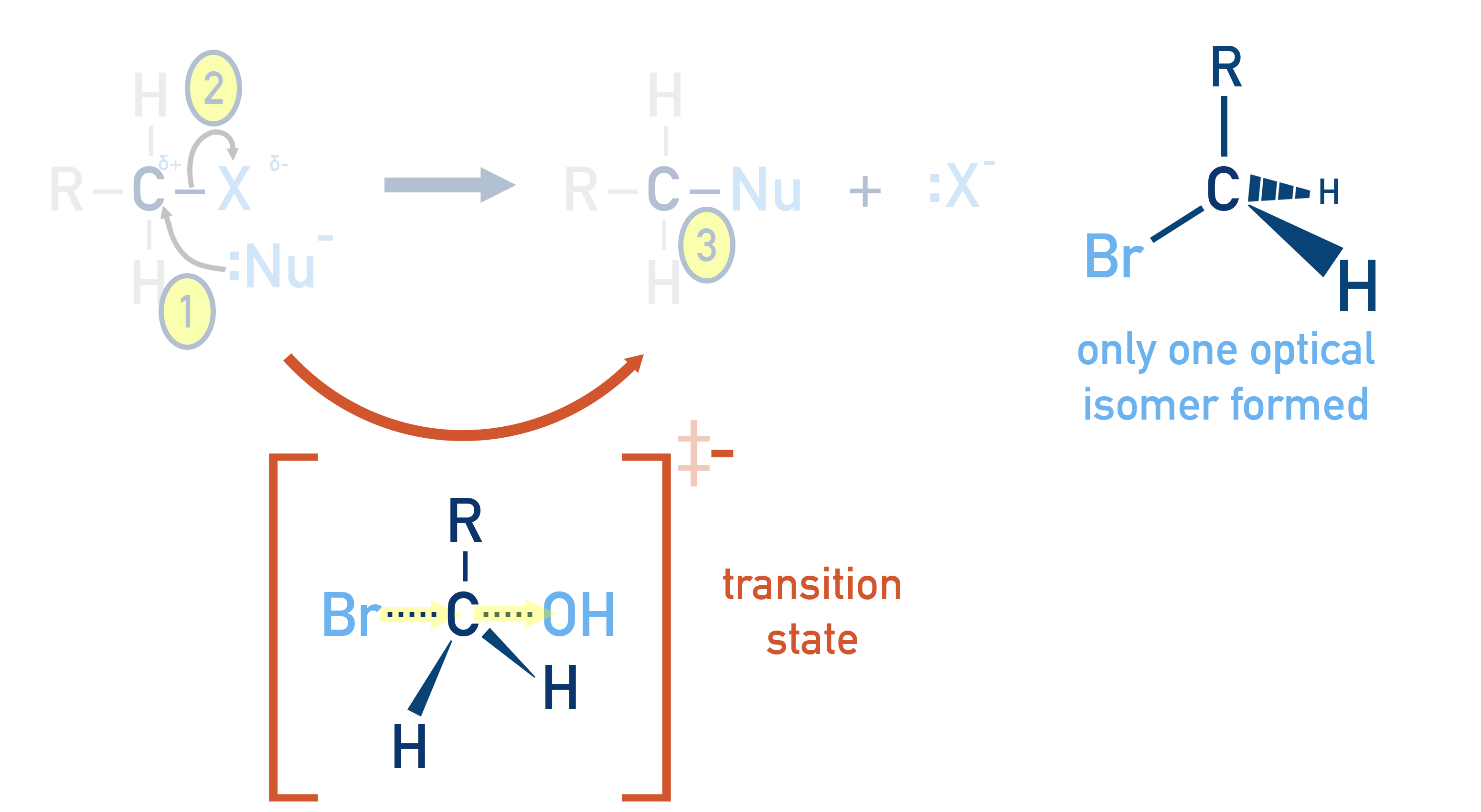
This produces only one stereoisomer (specifically an optical isomer), meaning the product mixture is optically active.
Elimination Reactions
Haloalkanes can undergo β-elimination in the presence of a strong base to form alkenes. This reaction competes with substitution under basic conditions.
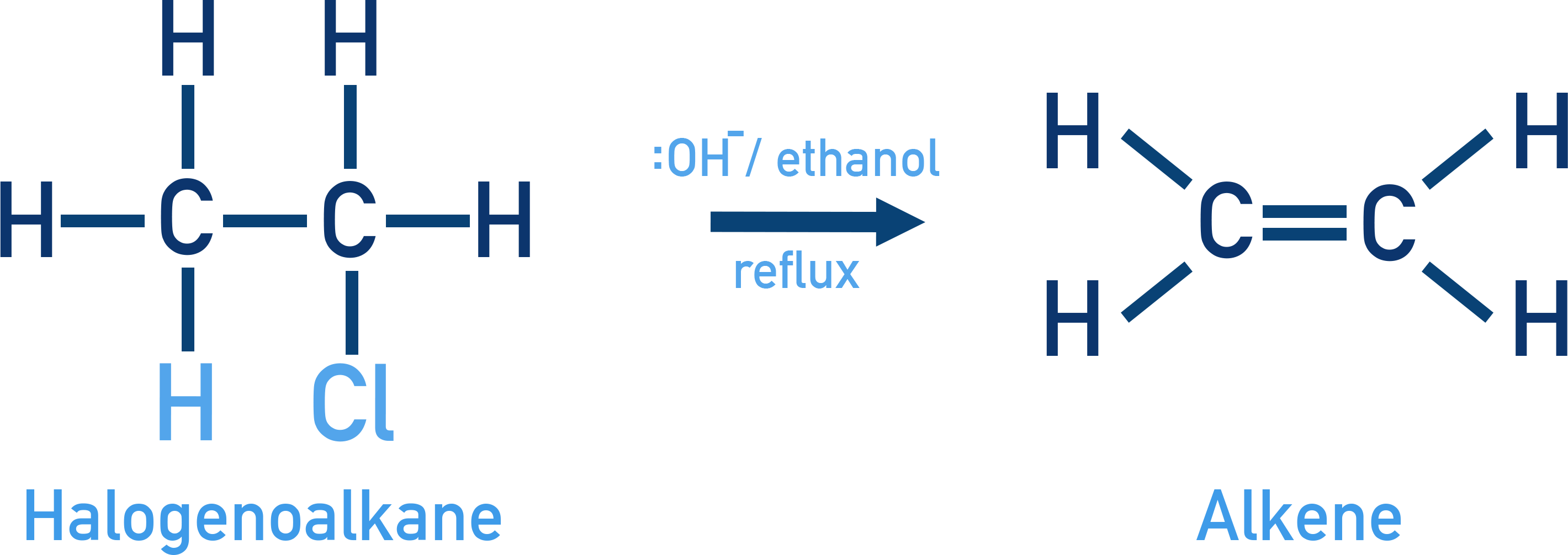
Mechanism:
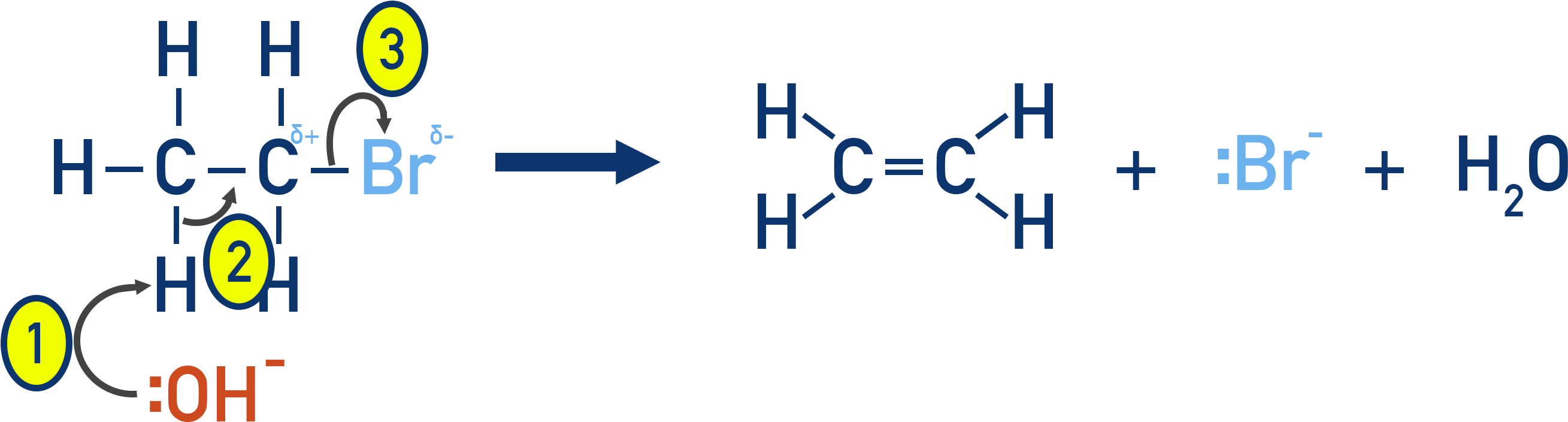
This mechanism is known as β-elimination because the hydroxide ion removes a hydrogen atom from the β-carbon – the carbon atom directly next to the α-carbon (which is bonded to the halogen).
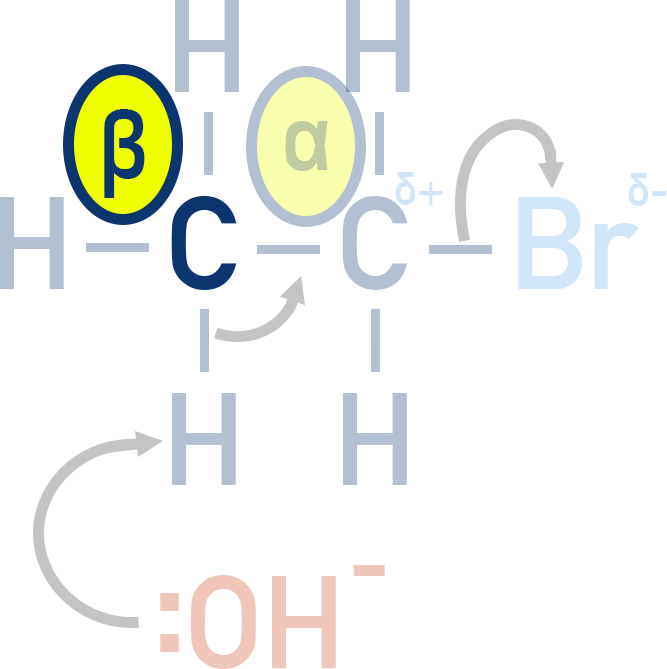
Zaitsev’s Rule: Where applicable, the more substituted alkene (with more alkyl groups on the double bond) is the major product.
- Influenced by:
- Type of alkyl halide (tertiary favours elimination)
- Strength of base
- High temperature
Example Dehydrohalogenation of 2-bromobutane
2-Bromobutane + alc. KOH → But-2-ene (major) + But-1-ene (minor)
Formation of Organo-Metallic Compounds
Haloalkanes (mainly chlorides, bromides, and iodides) react with certain metals to form compounds containing carbon–metal bonds. These are called organo-metallic compounds.
Grignard Reagents
Discovered by Victor Grignard in 1900. He developed alkyl magnesium halides (RMgX), now known as Grignard Reagents
Preparation: Formed by reacting a haloalkane with magnesium metal in dry ether:
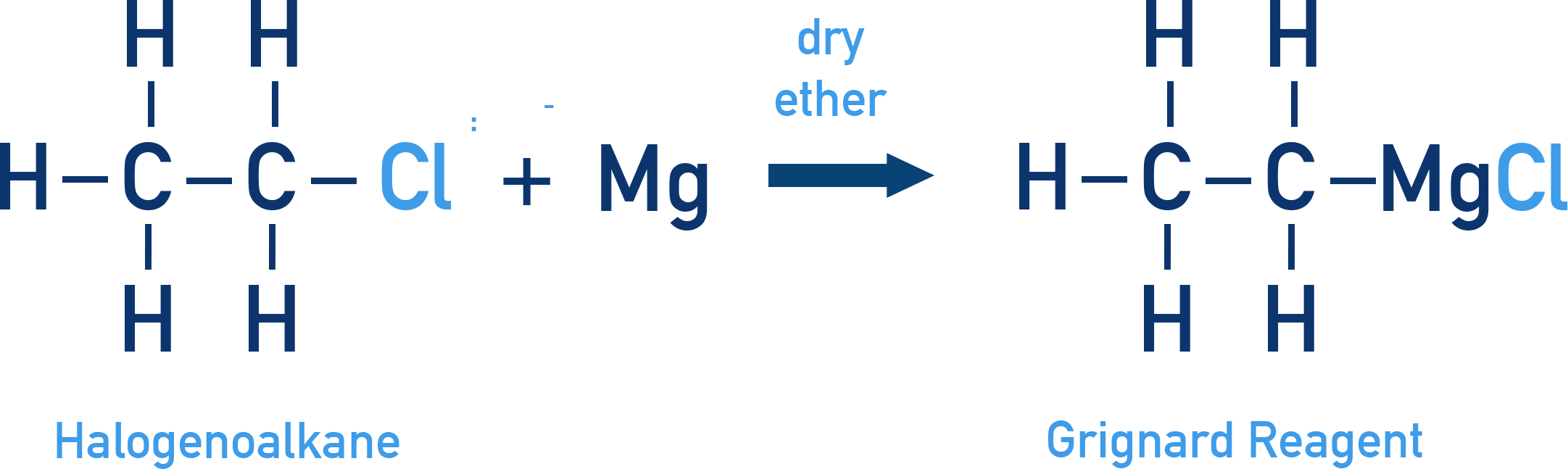
The C–Mg bond is covalent but highly polar, with carbon being partially negative (δ−). This means in reactions, the Mg–X bond is essentially ionic making Grignard reagents highly reactive. They can even react with weak acids like water or alcohols:
RMgX + H2O → RH + Mg(OH)X
Because of this, they must be handled in anhydrous (dry) conditions.
Wurtz Reaction
Haloalkanes react with sodium in dry ether to give hydrocarbons with double the number of carbon atoms:
2RX + 2Na → RR + 2NaX
This is known as the Wurtz reaction and is useful for forming longer-chain hydrocarbons.
Example Formation of ethane
CH3Br + 2Na + BrCH3 → CH3–CH3 + 2NaBr
Reactions of Haloarenes
Nucleophilic Substitution
Haloarenes (aryl halides) are much less reactive than haloalkanes toward nucleophilic substitution due to several stabilizing effects in the aryl system.
Why haloarenes are less reactive:
- Resonance effect
In haloarenes, the lone pair on the halogen interacts with the π-electrons of the benzene ring. - Hybridisation of carbon
In haloarenes, the halogen is bonded to an sp2-hybridised carbon (more electronegative, giving a stronger bond).
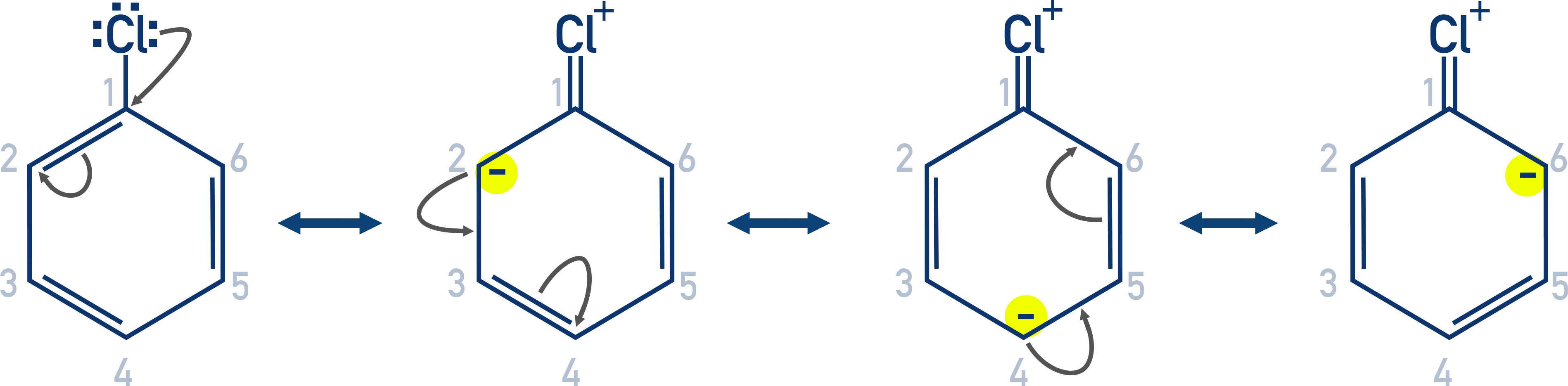
This delocalization creates resonance structures, giving partial double bond character to the C–X bond. As a result, breaking the C–X bond becomes more difficult.
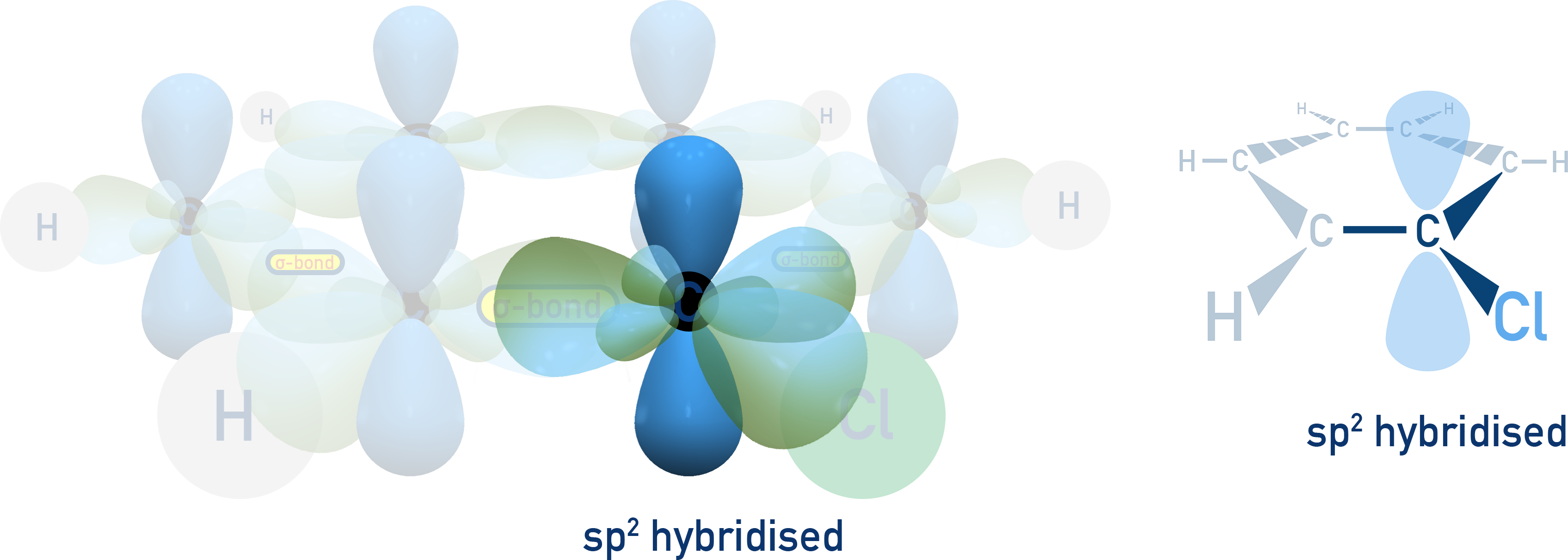
- In haloalkanes, it's bonded to an sp3-hybridised carbon (carbon atom is less electronegative, meaning a weaker bond).
- Instability of phenyl carbocation
Haloarenes cannot proceed via the SN1 mechanism. The phenyl carbocation that would form is unstable and not stabilized by resonance. - Electron-rich nature of the ring
The π-electron-rich benzene ring repels nucleophiles, making attack less favourable.

Bond length in haloalkanes (e.g. C–Cl = 177 pm) is longer than in haloarenes (C–Cl = 169 pm), making it easier to break.
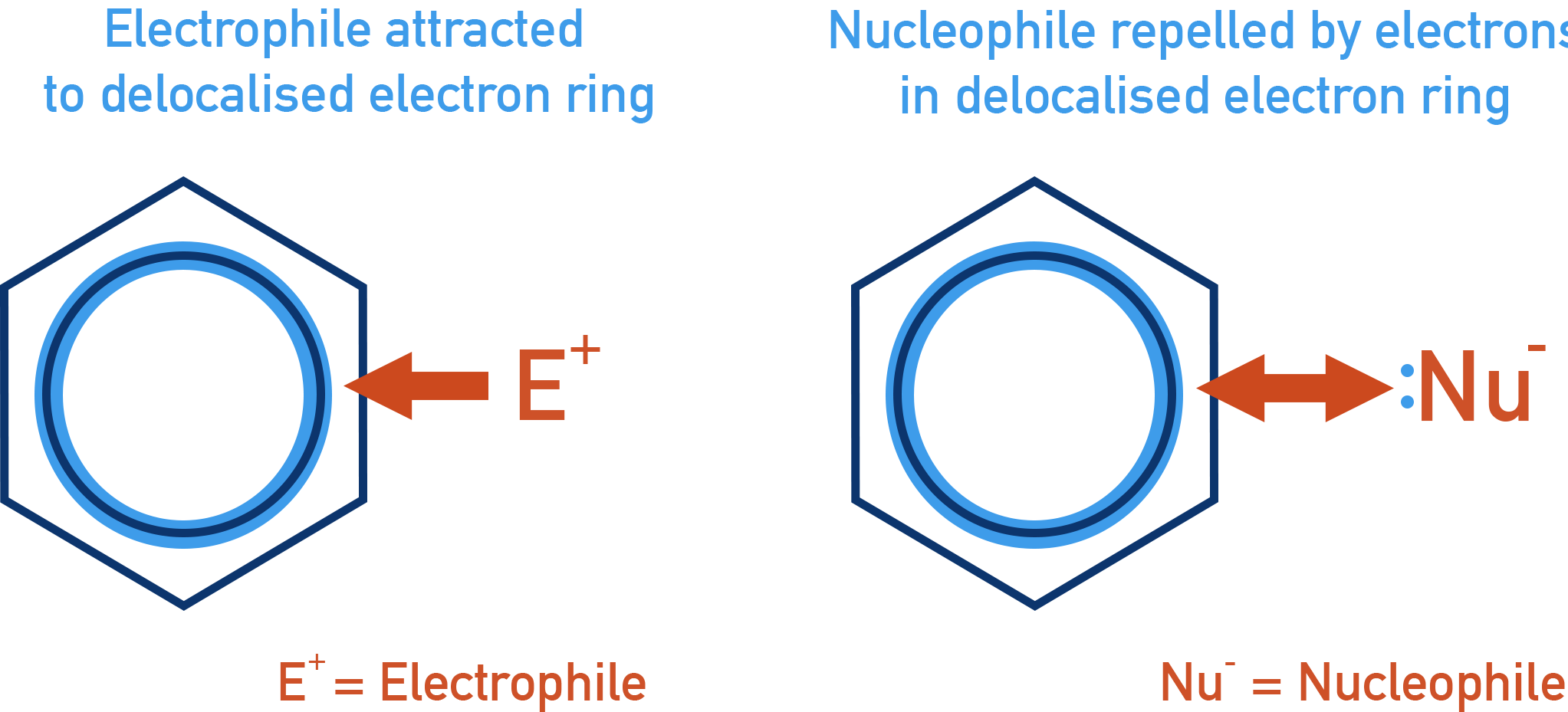
Nucleophilic Substitution via Hydroxide Ion (Replacement by OH−)
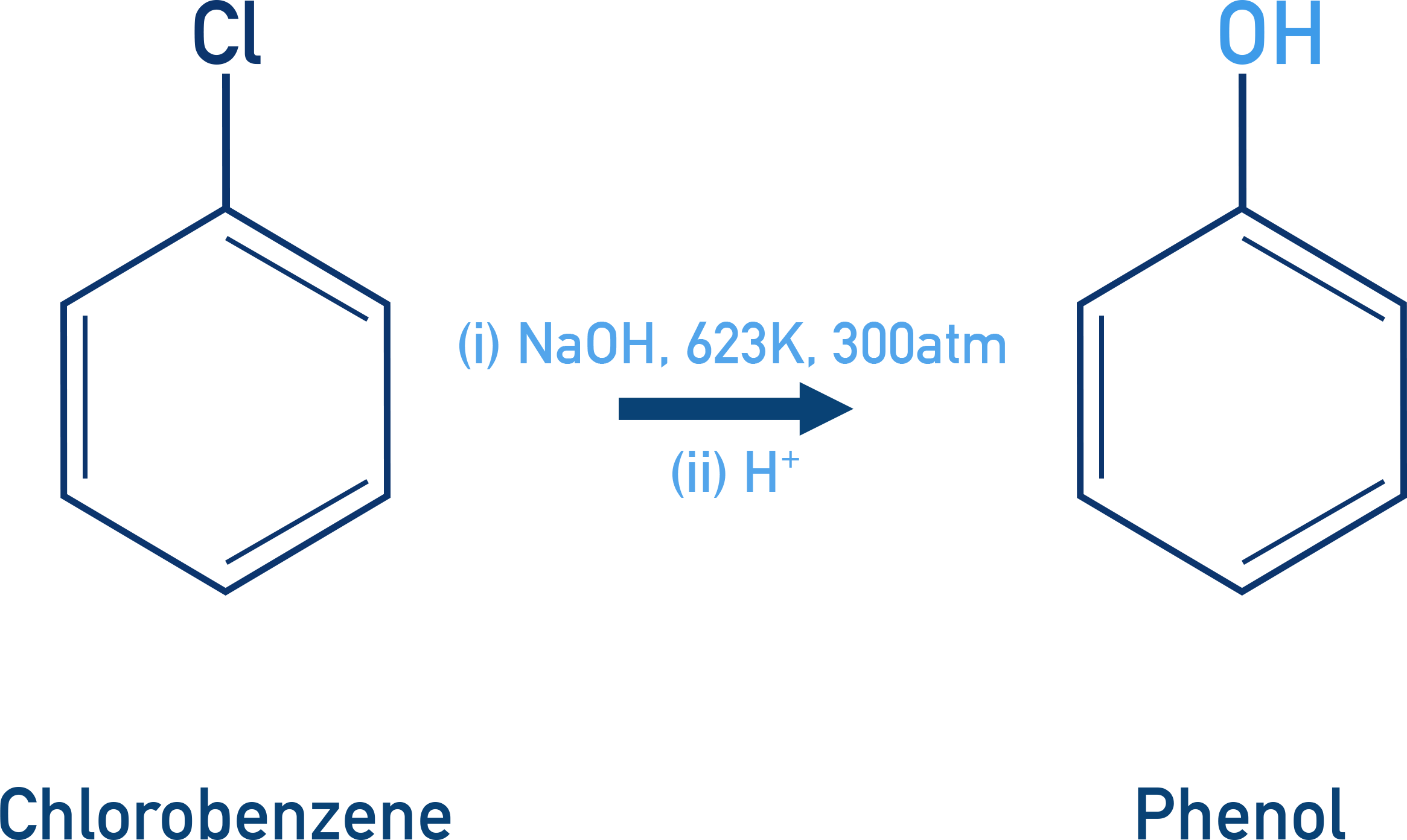
Chlorobenzene does not react easily with aqueous NaOH meaning harsh conditions are needed:
At 623 K and 300 atm, chlorobenzene can be converted to phenol:
Milder conditions can be used with –NO2 groups: The presence of electron-withdrawing groups like –NO2 at ortho or para positions increases reactivity.
Mechanism:
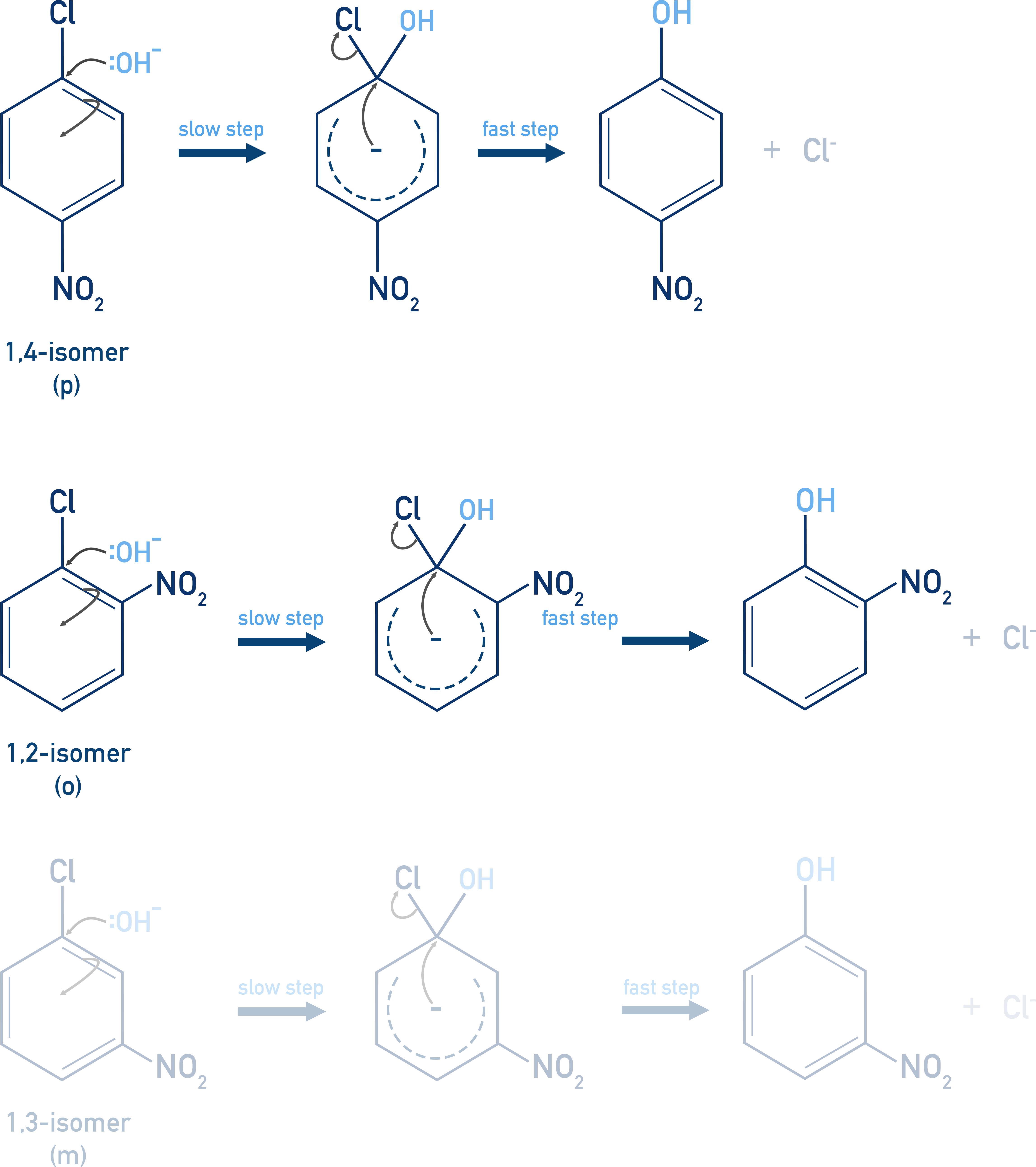
- Nucleophilic attack by OH− forming a negatively charged Meisenheimer complex (slow step)
- Elimination of Cl− to restore aromaticity (fast step)
This is favoured when electron withdrawing groups like –NO2 stabilize the intermediate through resonance. Meta-positioned –NO2 groups do not help, as they cannot stabilize the intermediate via resonance.
Haloarenes are less reactive toward nucleophiles than haloalkanes due to resonance and partial double bond character of the C–X bond.
Replacement by Hydroxyl Group
- Requires harsh conditions: high temperature and pressure.
- Electron-withdrawing groups (like NO2) at ortho/para positions facilitate the reaction.
Example Industrial conversion
C6H5Cl + NaOH (623 K, 300 atm) → C6H5OH + NaCl
Electrophilic Substitution Reactions
Despite being deactivating, halogens direct incoming electrophiles to ortho and para positions on the ring due to resonance.
Common Electrophilic Substitution Reactions:
Halogenation:
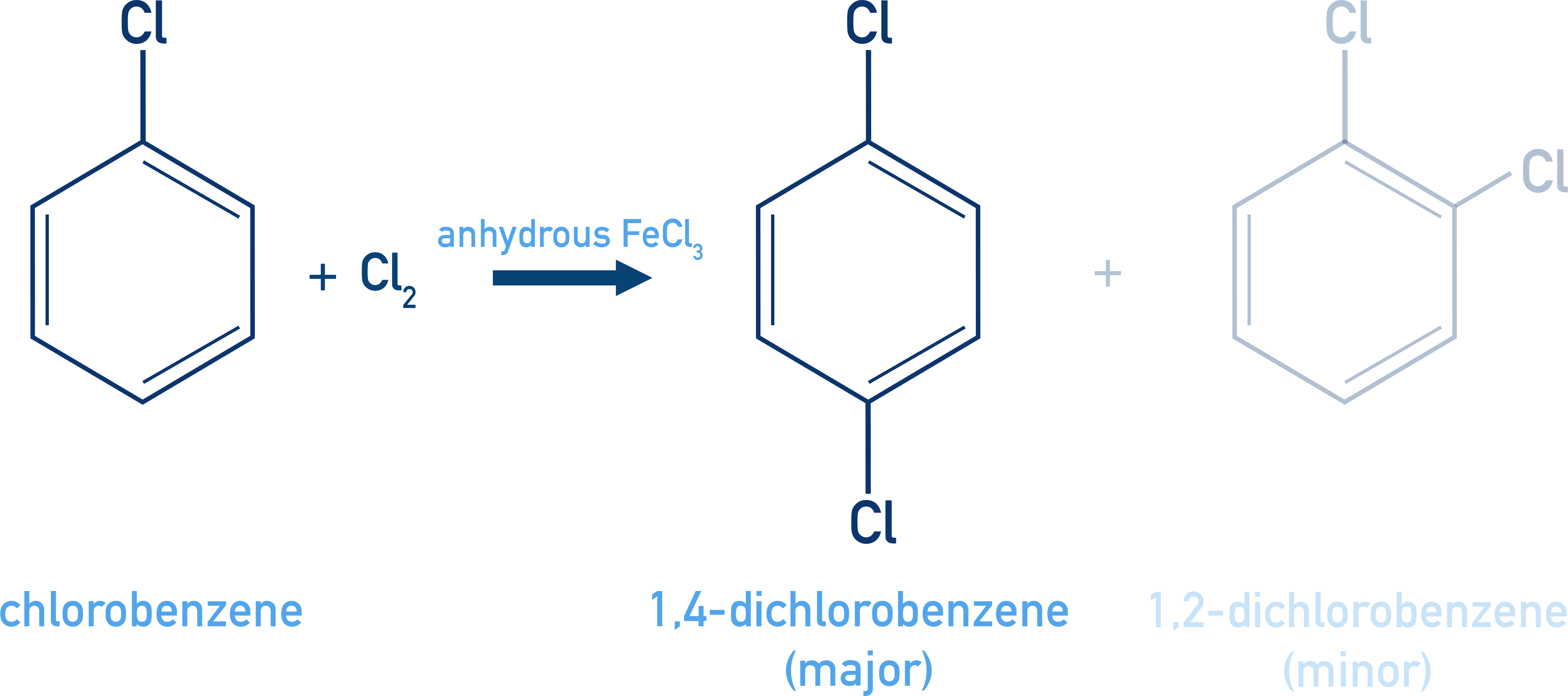
C6H5Cl + Cl2 (FeCl3) → o- and p-dichlorobenzene
Nitration:
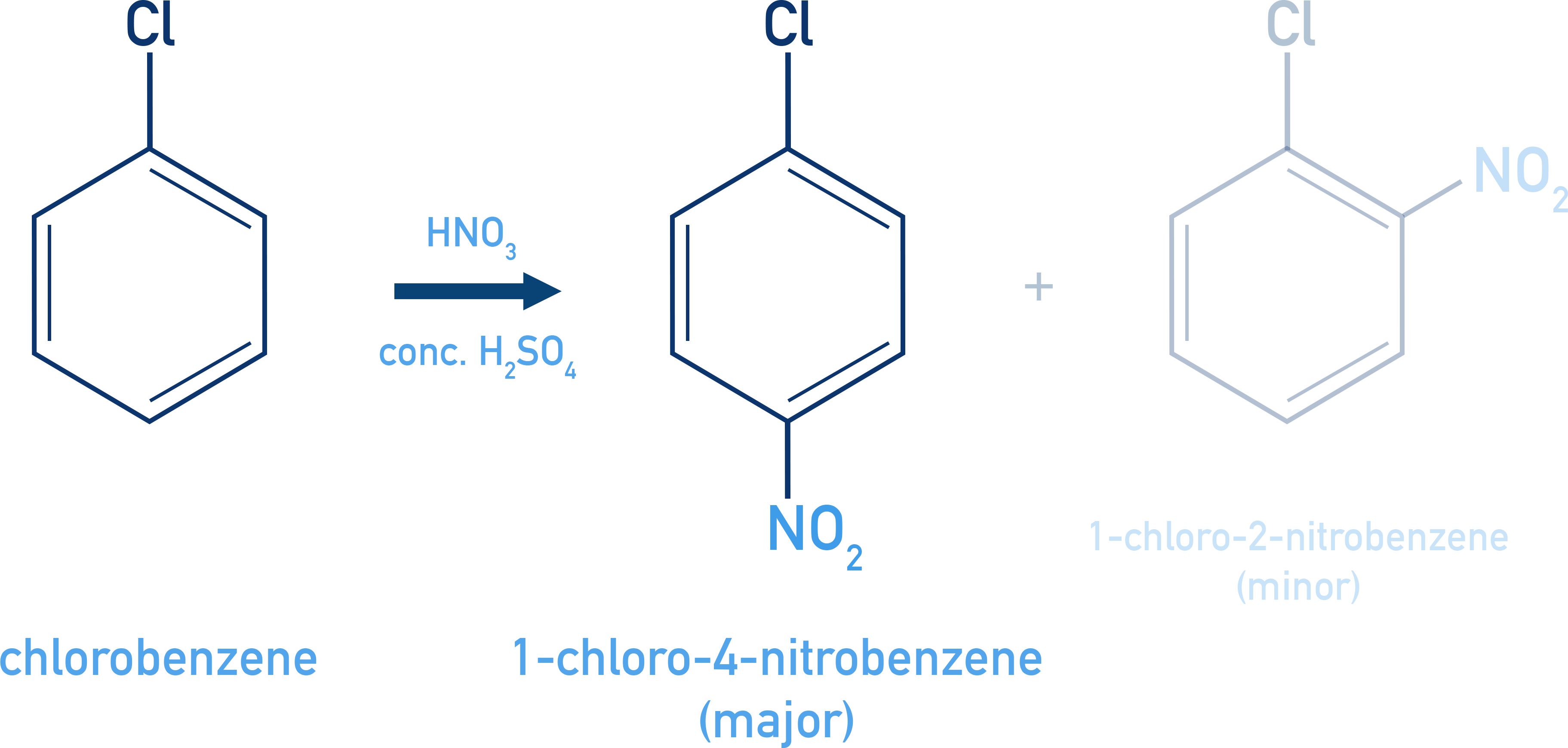
C6H5Cl + HNO3 (H2SO4) → o- and p-nitrochlorobenzene
Sulphonation:
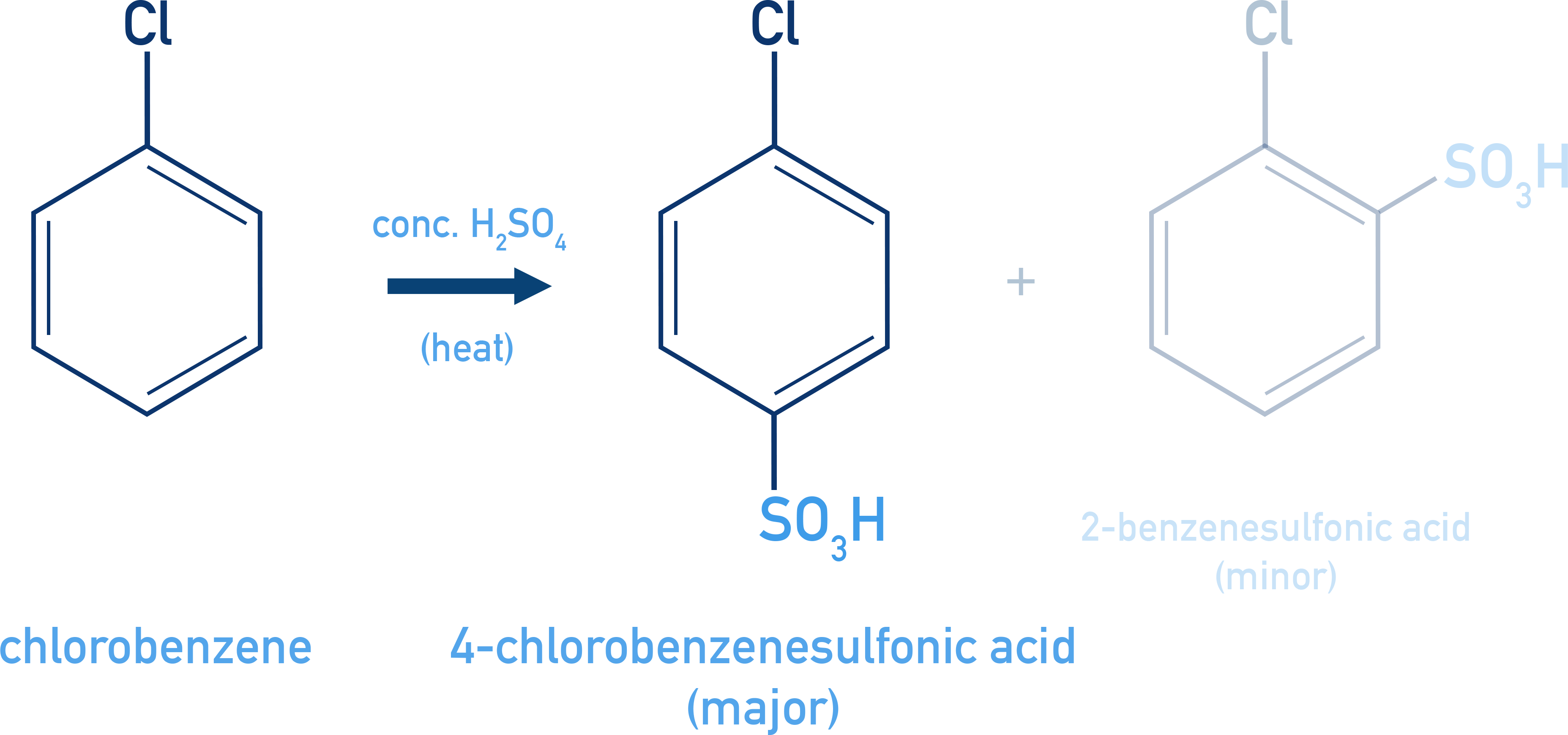
C6H5Cl + H2SO4 → o-/p-chlorobenzenesulphonic acid
Friedel–Crafts Alkylation:
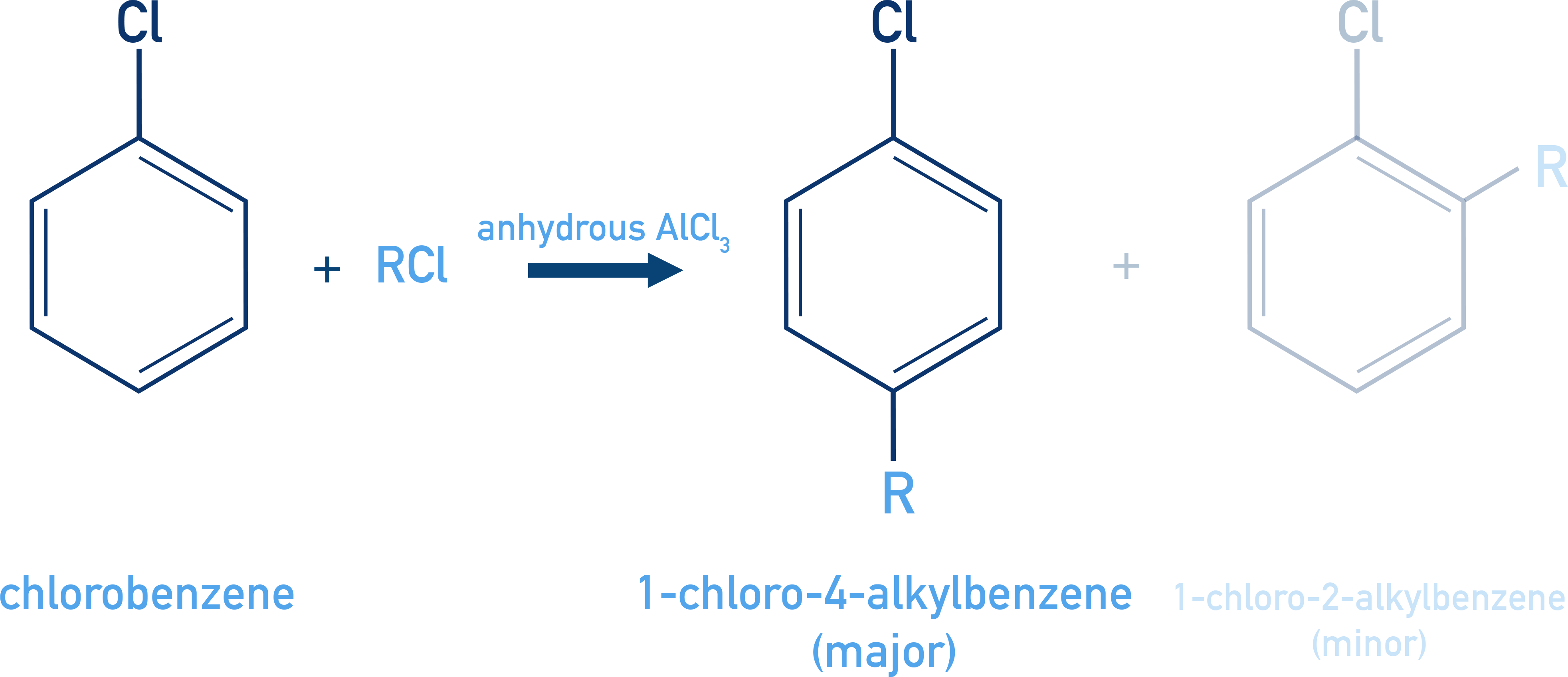
C6H5Cl + R–Cl (AlCl3) → o-/p-alkyl chlorobenzene
Friedel–Crafts Acylation:
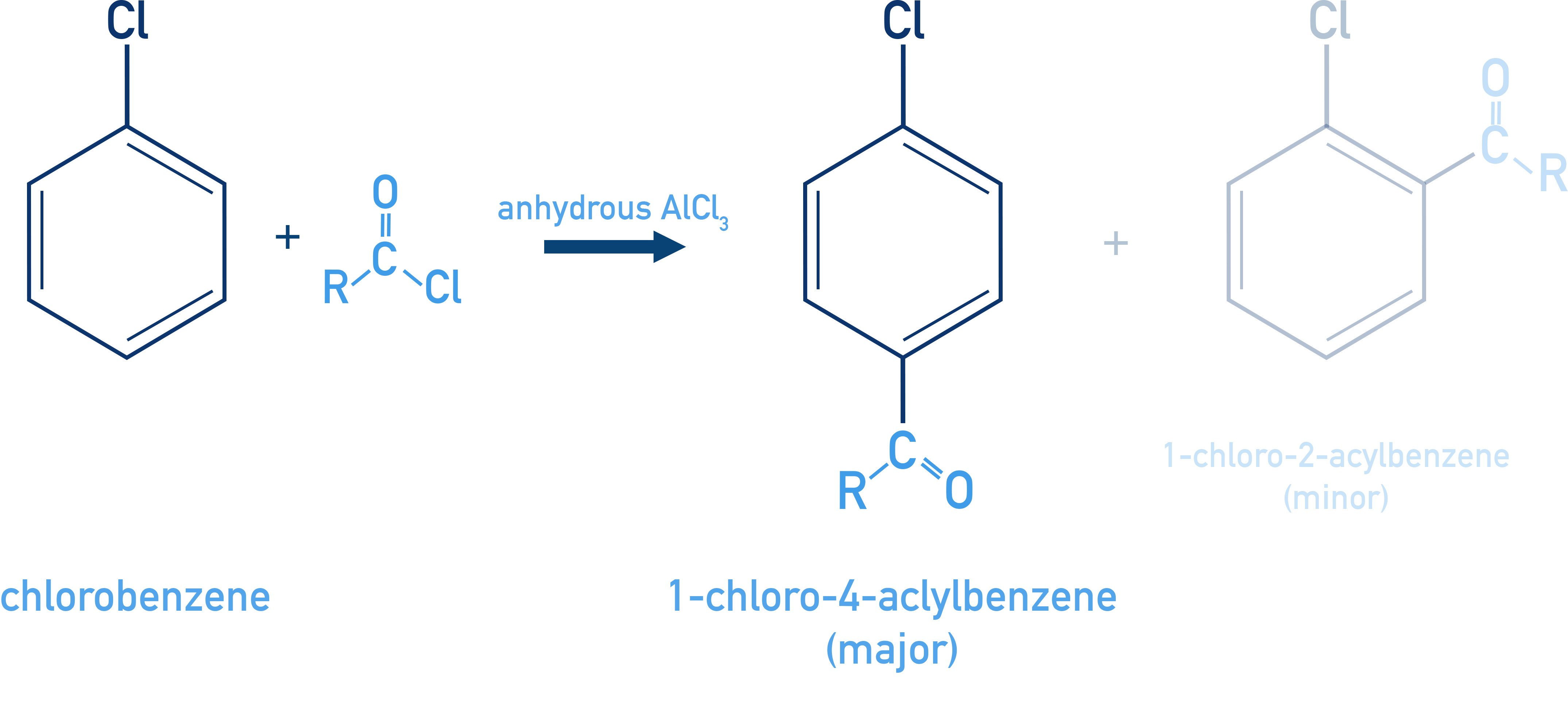
C6H5Cl + RCOCl (AlCl3) → o-/p-acyl chlorobenzene
Reactions with Metals
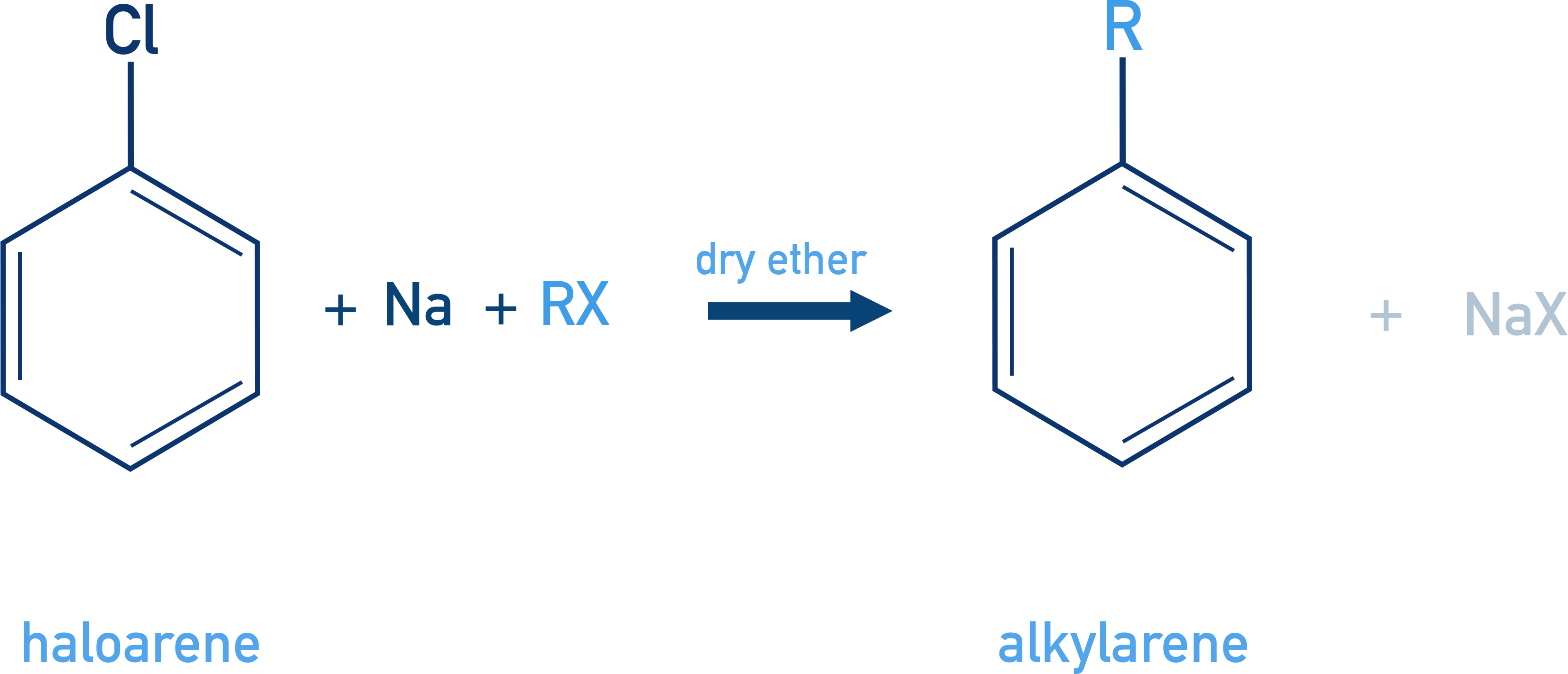
Wurtz–Fittig Reaction
Used to form alkylbenzenes by reacting haloarenes and haloalkanes with sodium in dry ether.
C6H5X + R–X + 2Na → C6H5–R + 2NaX
Example Toluene formation
C6H5Br + CH3Br + 2Na → C6H5CH3 + 2NaBr
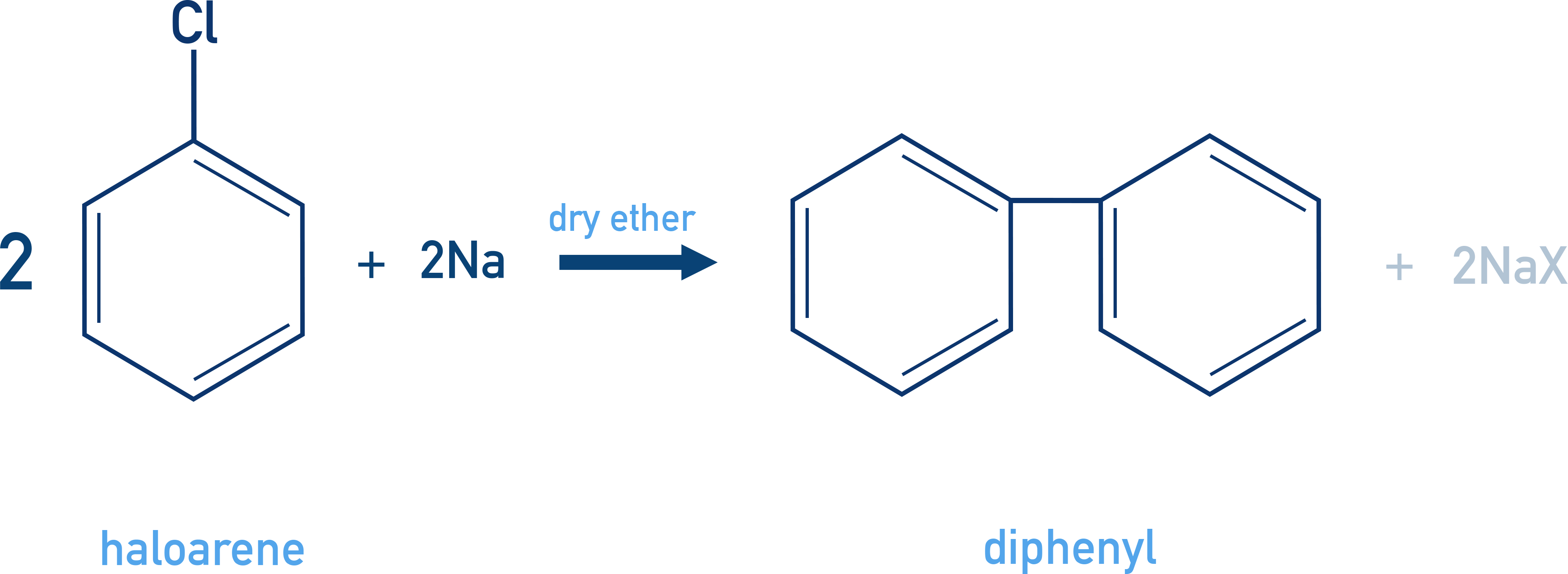
Fittig Reaction
Couples two aryl halides to form biaryl compounds like biphenyl.
2 C6H5X + 2Na → C6H5–C6H5 + 2NaX
Example Biphenyl synthesis
C6H5Br + Na → Biphenyl + NaBr
Summary
- SN1 vs SN2 reactions depend on the substrate structure and solvent.
- Haloalkanes readily undergo nucleophilic substitution and elimination while haloarenes generally do not because of resonance.
- Haloarenes prefer electrophilic substitution and direct to ortho and para positions.
- Metal-coupling reactions and Grignard formation are valuable synthetic tools for building larger frameworks.