
Benzene Structure and Naming Aromatic Compounds
Quick Notes
- Structure and Bonding in Benzene
- Benzene has a cyclic, planar hexagonal structure.
- It features delocalised π-electrons formed by p-orbital overlap.
- All C–C bond lengths are equal; intermediate between single and double.
- Delocalised π-electrons (ring of delocalised electrons from the delocalisation of p electrons) increase benzene’s stability compared to the theoretical cyclohexa-1,3,5-triene (Kekulé model). Evidence for the delocalised model includes:
- Bond length measurements: all C–C bonds equal.
- Hydrogenation enthalpy: less exothermic than expected.
- Benzene resists addition reactions, favouring substitution.
- Naming Aromatic Compounds
- Simple groups like methyl, nitro, or chloro are given as prefixes, with positions numbered to give the lowest possible values in the name.
- Benzene as a functional group is named as ‘phenyl’.
Full Notes
Benzene is an unsaturated hydrocarbon with the molecular formula C6H6.
Bonding in Benzene
The Kekulé model suggests alternating double and single C–C bonds.
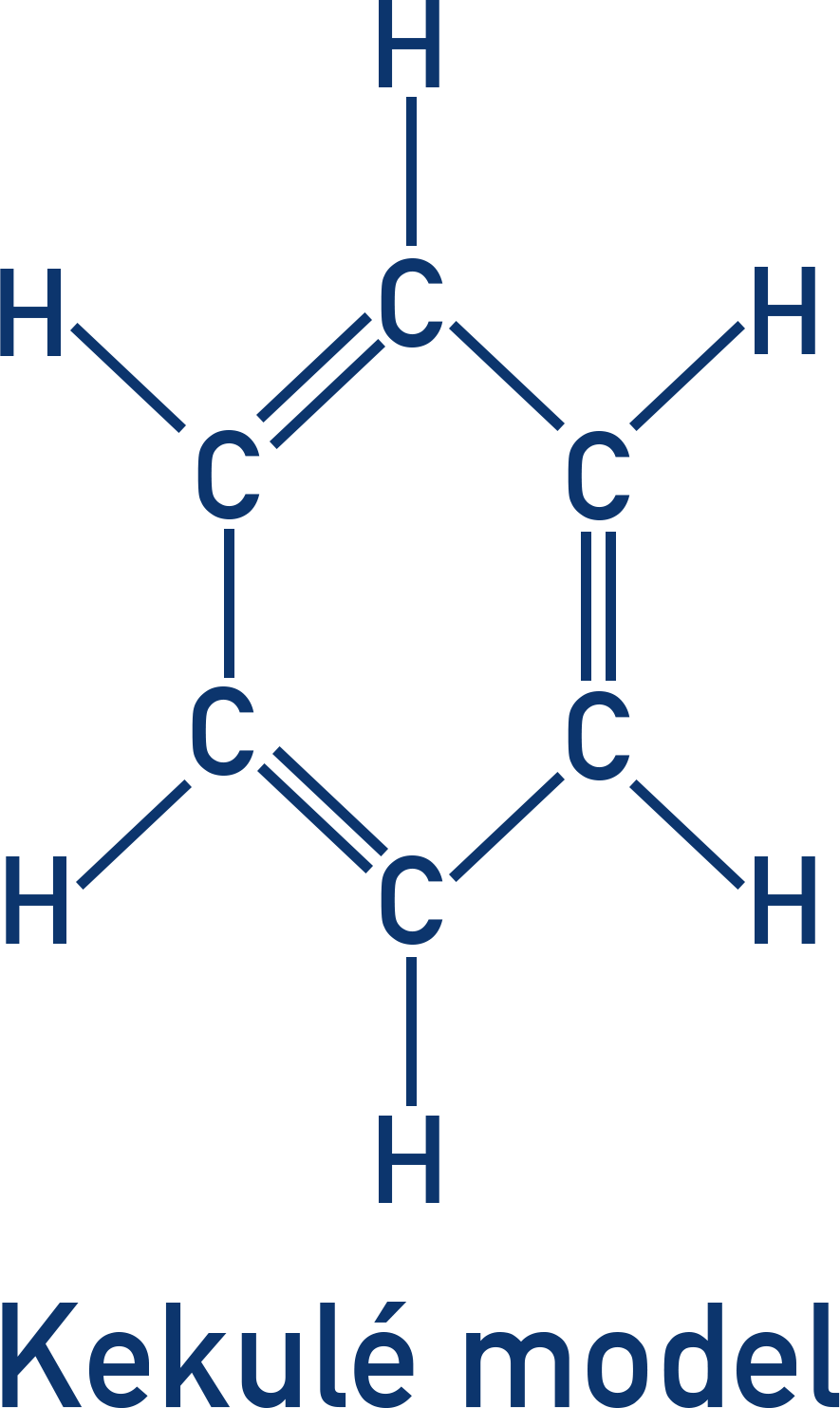
However, X-ray diffraction shows that all C–C bonds are the same length (intermediate between single and double), and enthalpy changes of hydrogenation show benzene is more stable than expected.
Because of this we now use another model, called the delocalised model.
There is a delocalised system of π electrons, formed by the sideways overlap of unbonded p orbitals from each carbon atom.
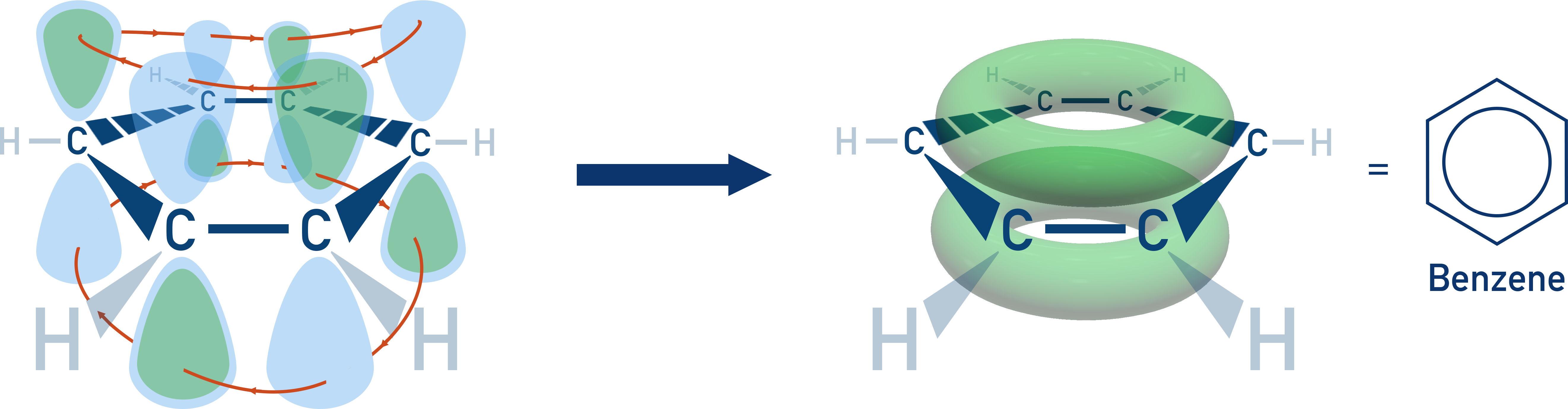
These six p electrons form a continuous cloud of electrons above and below the plane of the carbon atoms, leading to uniform bond lengths and a more stable structure.

Make sure you can explain why we now propose the delocalised electron model of benzene – evidence of equal bond lengths, hydrogenation enthalpies and a tendency for substitution rather than addition reactions. Remember the theoretical molecule cyclo-1,3,5-triene (Kekulé model) doesn’t actually exist.
Evidence for Delocalisation: Bond Lengths
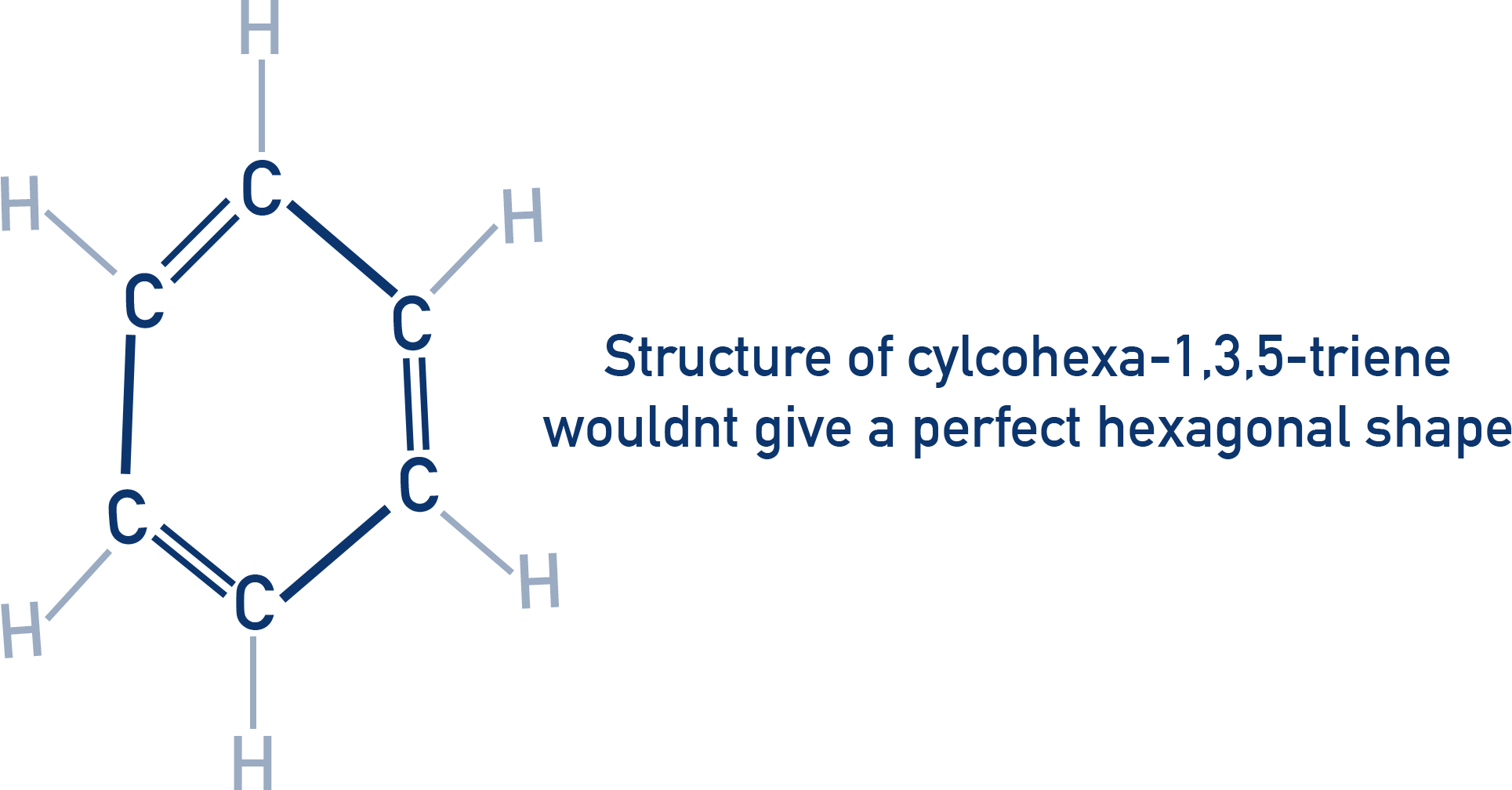
All C–C bond lengths in benzene are equal (~0.140 nm). This is between the length of a single bond (0.154 nm) and a double bond (0.134 nm).
As a result, benzene’s structure cannot be represented accurately by alternating single and double bonds (as in the theoretical Kekulé model of cyclohexa-1,3,5-triene).
Evidence for Delocalisation: Enthalpies of Hydrogenation
Enthalpies of hydrogenation show that benzene is more stable than expected – especially when compared to the theoretical cyclohexa-1,3,5-triene structure.
Less energy is released when benzene is hydrogenated and turned into cyclohexane than would be predicted for cyclohexa-1,3,5-triene.
Hydrogenation of cyclohexene (one C=C bond): ΔH = −120 kJ mol−1
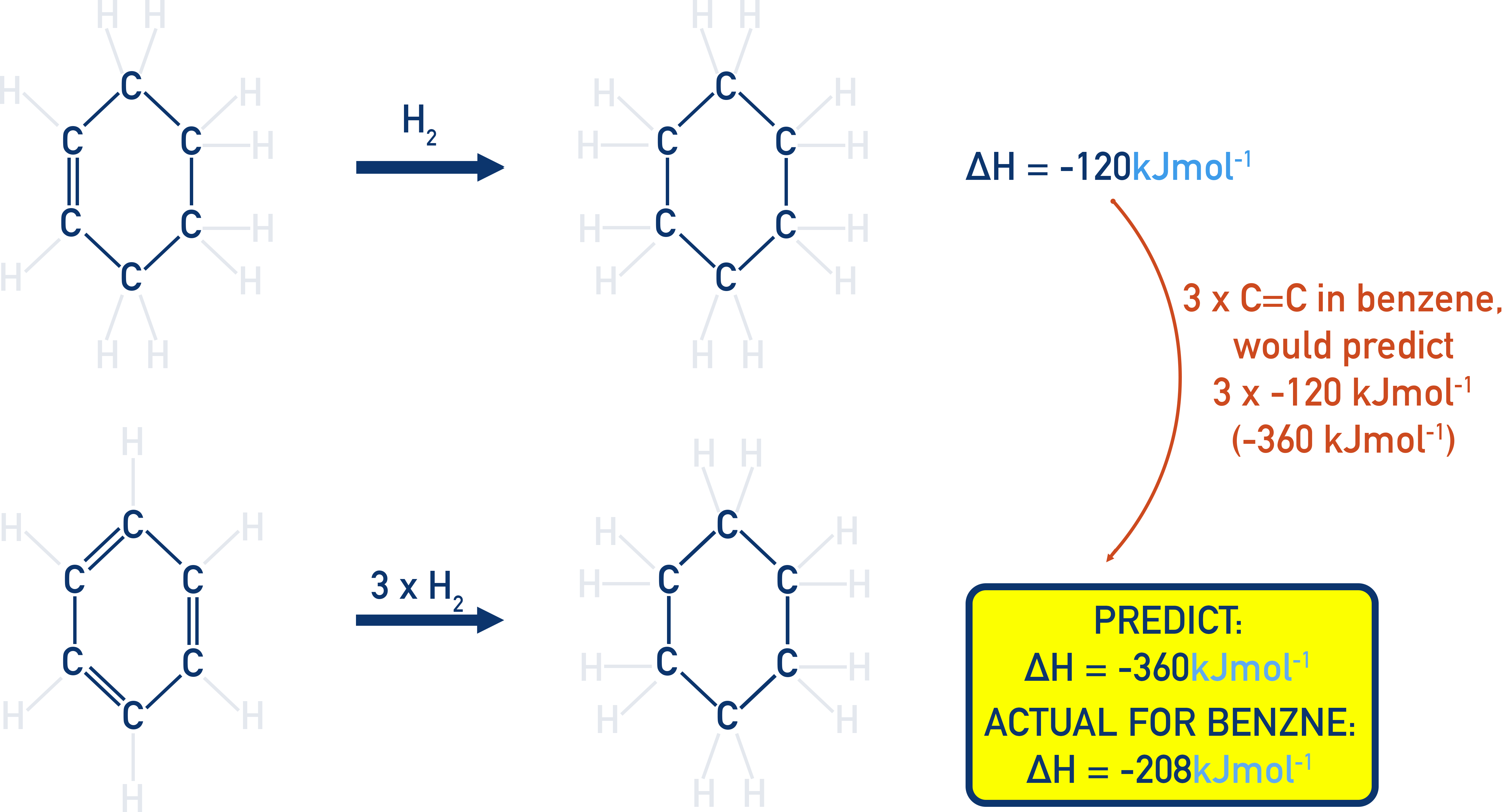
Expected hydrogenation of cyclohexa-1,3,5-triene (three C=C bonds): ΔH = −360 kJ mol−1
Actual hydrogenation of benzene: ΔH = −208 kJ mol−1
This less exothermic enthalpy change for benzene indicates it is more stable than expected. This is now explained by delocalisation energy.
Reactivity of Benzene
Benzene is less reactive than alkenes because of its electron delocalisation, which spreads the π-electron density evenly across the ring. This delocalisation makes it harder to attract electrophiles.
Naming Aromatic Compounds
Substituted benzenes are named using IUPAC rules: simple groups like methyl, nitro, or chloro are given as prefixes, with positions numbered to give the lowest possible values.
For Example: with phenols
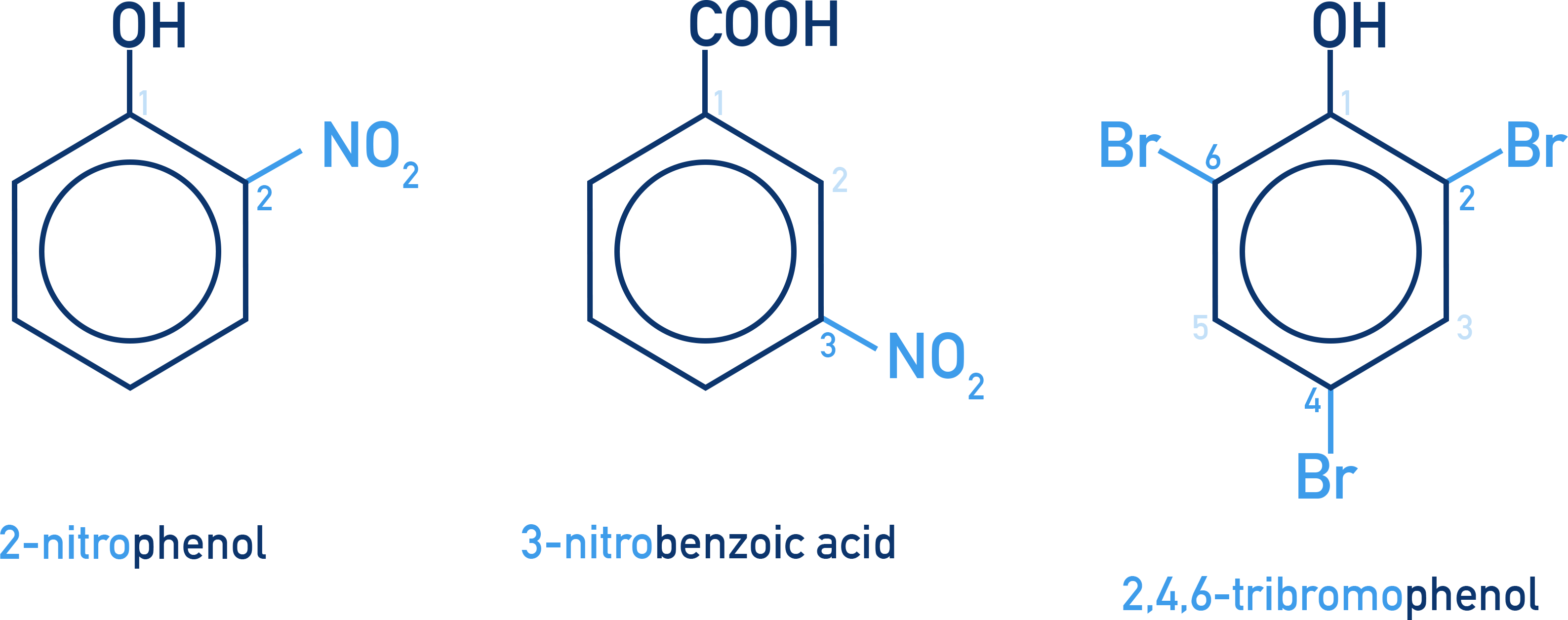
2-nitrophenol: A nitro group at position 2 relative to the hydroxyl group.
3-nitrobenzoic acid: Nitro group on carbon 3 of the benzene ring with a carboxylic acid group at position 1.
2,4,6-tribromophenol: Bromine atoms at positions 2, 4, and 6; hydroxyl group at position 1.
Summary
- Benzene is cyclic and planar with a delocalised π-system that gives equal C–C bond lengths.
- Evidence for delocalisation includes bond length equality and less exothermic hydrogenation enthalpy than expected.
- Benzene’s stability explains its preference for substitution over addition.
- Aromatic compounds are named with substituent prefixes and the phenyl group when benzene is a substituent.