Video Tutorial NMR Spectroscopy
Quick Notes NMR Spectroscopy
- Nuclear Magnetic Resonance (NMR) spectroscopy shows how many unique environments there are in a compound and how they interact with one another.
- 13C NMR – shows unique carbon environments.
- 1H NMR – shows unique hydrogen (proton) environments.
- Sub-atomic particles spin in two directions (+½ or -½), creating a very weak magnetic field.
- Nuclei with an odd number of sub-atomic particles have an overall spin.
- Nuclei are placed in an external magnetic field (B0), forcing them to have a +½ spin. Energy from radio waves is used to force the nuclei to ‘flip’ their magnetic fields and have a -½ spin, making them align against (resonate) the external magnetic field.
- The energy required is measured and displayed on a spectra as absorptions, where each peak shows a different nuclei environment in a molecule.
- NMR samples are compared to a standard (usually TMS).
- All the carbon and proton environments in TMS are the same, showing one absorption with a frequency ‘defined’ as zero, 0.
- Chemical shift refers to the difference between the absorption in TMS (0) and in the nuclei of a sample compound.
- The amount of energy required to resonate the nucleus’ magnetic field changes depending on the electrons around the nucleus and the environment of the nucleus in a molecule.
- A nucleus with a lower electron density is easier to resonate – its absorbance is downshifted (further away from 0).
- A nucleus with a higher electron density is harder to resonate – its absorbance is upshifted (closer to 0).
- Spin-spin coupling is the interaction of protons on adjacent, non-equivalent carbon atoms, which causes split peaks on a spectra.
- The number of times a peak can be split is dependent on the number (n) of protons bonded to adjacent, non-equivalent carbon atoms.
- n+1 is the number of times a peak will be split.
Full Notes NMR Spectroscopy
Nuclear Magnetic Resonance (NMR) spectroscopy is a powerful technique widely used in the analysis of organic compounds. At its simplest, NMR is used to find out how many unique environments there are in a compound and how, with more advanced analysis, these environments interact with one another.
There are two main types of NMR spectroscopy: carbon (13C NMR) and proton (1H NMR). Carbon NMR is generally the easier of the two methods to analyse as it simply shows how many unique carbon environments there are in a compound.
Proton NMR follows the same basic theory as carbon NMR, except it’s analysing the proton (hydrogen) environments within a compound. With high resolution NMR, how the proton environments interact with each other can give information about how the environments are arranged in the compound, allowing potential structures to be predicted. These ‘interactions’ arise due to a phenomenon called ‘spin coupling’. Spin coupling is commonly the area of NMR students find most demanding, and analysing spectra (graphs produced during NMR analysis) is a skill that takes time and practice to build.
Background Theory
A-Level chemistry students are not required to understand the complete background theory to NMR spectroscopy. However, knowing how the process works helps enormously with interpreting spectra data.
Sub-atomic particles possess a characteristic called ‘spin’. This spin, for A-level purposes, can be thought of as the particle spinning on its axis (rather like planet Earth ‘spinning’). There are only two directions a particle can spin in; these directions are referred to as +½ and -½.
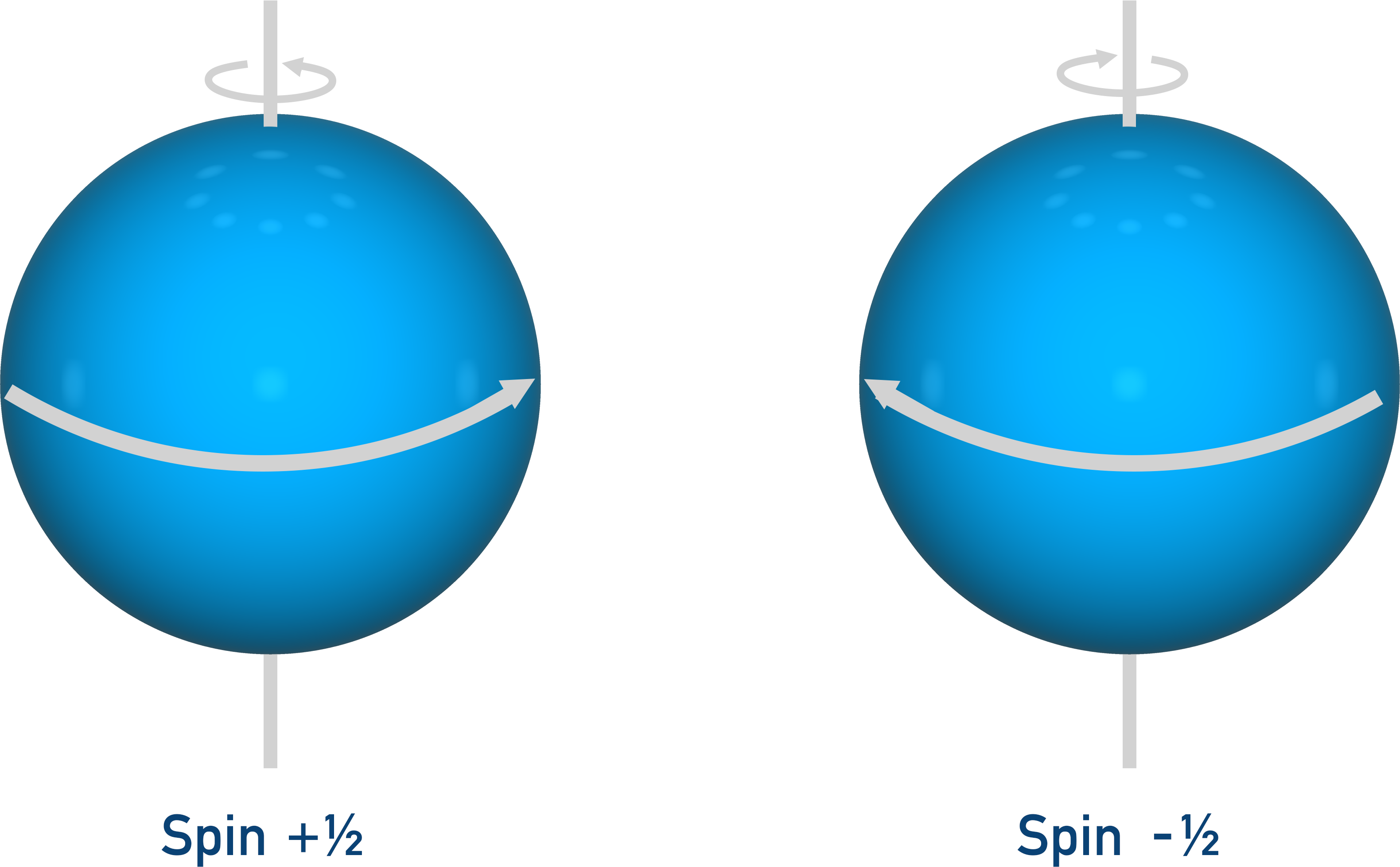
With NMR, it’s the nucleus of an atom we are interested in. In a nucleus with two particles of opposite ‘spin’ the particles will cancel each other out, so the nucleus will have no overall spin. A nucleus of three particles will always end up having an overall spin as only two particles can cancel out. Carbon-13 and hydrogen nuclei are used in NMR because their nuclei have an overall spin.
A nucleus with an overall spin is almost ‘wobbling’ on its axis (called angular momentum). Rather like a spinning top trying to stay upright.
When a charged particle spins, or rotates, a very weak magnetic field is created. So, a spinning nuclei (positively charged) with an odd number of sub-atomic particles will have a very weak magnetic field. When this happens, the nucleus can be thought of as a small ‘bar magnet’.
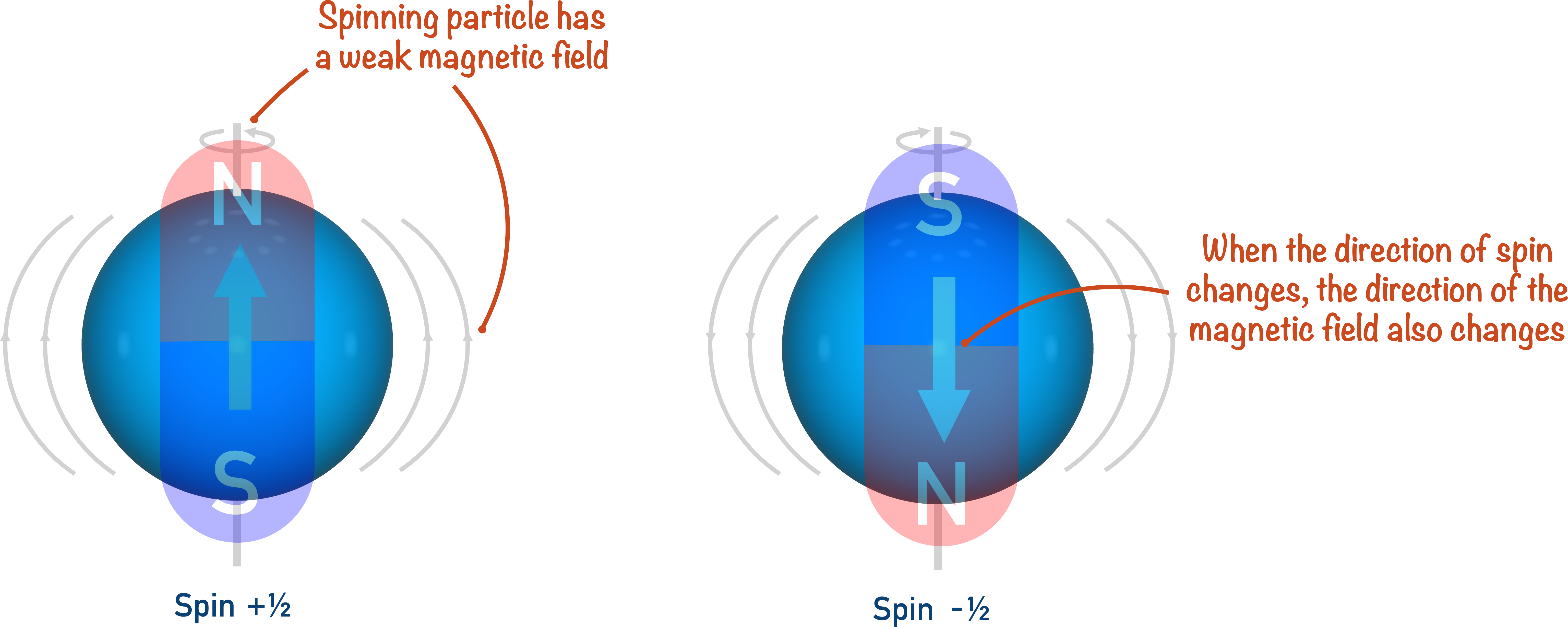
In a similar way to a bar magnet, the magnetic field created by the nucleus has a direction. It is the purpose of NMR spectroscopy to ‘flip’ this direction and, in very simple terms, compare the amount of energy required to do this for different nuclei environments.
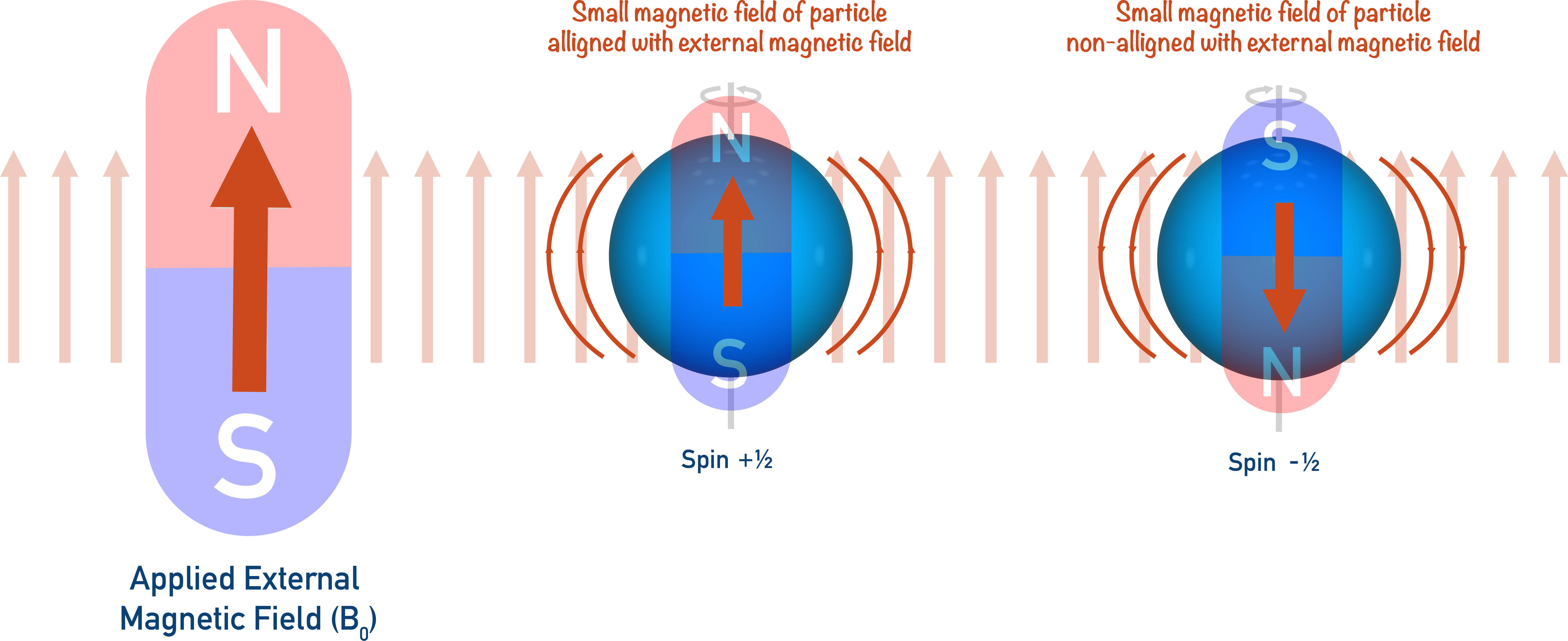
Imagine putting small bar magnets in a box surrounded by a larger magnet (an ‘external magnetic field’). The small bar magnets will all flip around – ‘aligning’ with the applied magnetic field.
If you wanted to ‘flip’ a small bar magnet again, you would have to use energy to force it to go against the external magnetic field. (A bit like trying to force two magnetic south poles together).
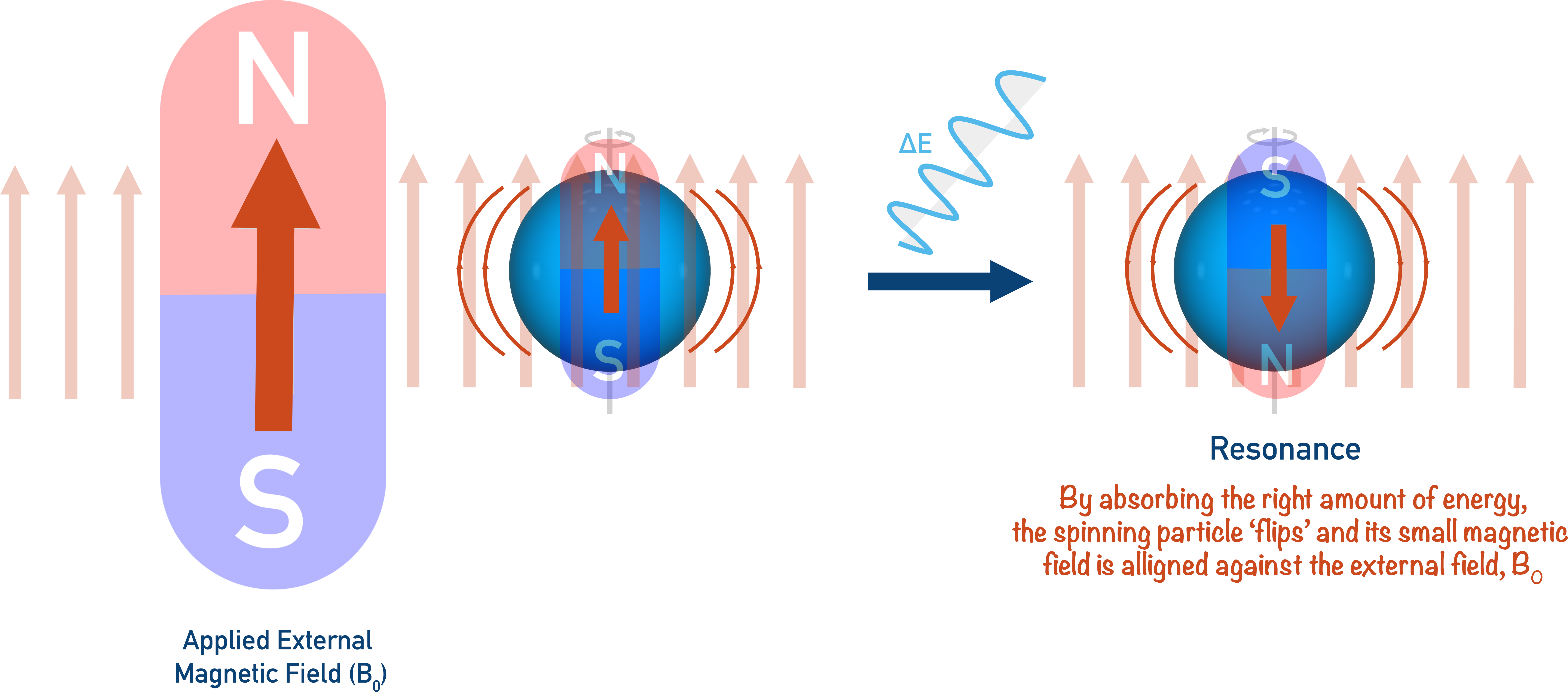
In NMR spectroscopy, it’s the same idea: a spinning nucleus acts as a small bar magnet, and an applied external magnetic field is applied (shown as B0).
The external magnetic field forces the nucleus to spin a particular direction (+½). By providing just the right amount of energy, the spinning nucleus can be forced to spin in the opposite direction (-½). When this happens, we say ‘resonance’ has occurred and the nuclei now has a magnetic field direction that is against the applied magnetic field (B0).
By changing the strength of the applied magnetic field, the energy required to change the spin of the nucleus can be changed and it can be made easier or harder to force the nucleus to reach resonance.
The energy used to ‘flip’ nuclei and cause resonance comes from radio waves with a set frequency. A sample is bombarded with radio waves (all with the same frequency) and a detector detects whether they have been absorbed or have passed though.
The radio waves will only be absorbed if the nuclei are resonating, otherwise they will pass straight through and be detected on the other side. By slowly changing the strength of the applied magnetic field (B0), eventually the energy from the waves will be just right to force the nuclei to resonate. At this point, the waves will be absorbed by the nuclei and won’t be detected in the machine. This is how we can know resonance has been achieved.
The relevant bit to chemists is the fact that nuclei are surrounded by electrons, each also spinning with their own magnetic fields. These small magnetic fields effectively ‘shield’ the nucleus from the external magnetic field. The amount of energy required to ‘flip’ the spin of the nucleus changes depending on the electrons around the nucleus and the environment of the nucleus in a molecule.
The lower the electron density around a nucleus, the easier it is to resonate the nucleus. The higher the electron density around a nucleus, the harder it is to resonate the nucleus.
Carbon and hydrogen nuclei within a molecule will be shielded differently if they are in different environments. Meaning, for a specific frequency radio wave, different strengths of applied magnetic field (B0) are required to flip their spin. These energies show on a spectra as absorptions, and the number of absorptions within a spectra indicates the number of different nuclei environments (for one type of nuclei) in a molecule.
Chemical Shift
To enable a comparison of nuclei environments within a compound, a chemical standard is used – usually TMS (Tetramethylsilane). All the carbon and proton environments in TMS are the same, meaning there is only one absorption for both the carbon and proton NMRs.
The frequency these absorbances occur at is ‘defined’ as zero. This means when a sample is analysed and the absorbances of different nuclei (carbon or proton) are measured, they are being compared to the absorbances of the same type of nuclei in TMS.
Chemical shift refers to the difference between the absorption in TMS and in the nuclei of a sample compound.
Carbon and proton nuclei in TMS have a high electron density around them, so there is a high level of shielding.
Alternatively, a proton in an OH group has a very low electron density around it, due to high electronegativity of the oxygen compared to the hydrogen. As a result, the proton in the OH group will absorb a different energy to the protons in TMS – measured as a ‘shift’ from 0 and plotted on a spectra.
The lower the electron density around a nucleus, the lower the energy required to ‘flip’ its magnetic field, and the further away from the standard (TMS) 0 peak it will be. The absorption is said to be ‘downshifted’.
Certain types of environment (for example, OH) will always have absorbances at similar shifts, enabling types of nuclei environment to be predicted from the position of a chemical shift on a spectra. Very small changes in electron environments change the exact chemical shift, so a ‘range’ of shifts are usually given for a particular type of environment. For OH, this range is usually 0.5 to 5.
Spin-spin Coupling
Perhaps the most useful, but also most confusing, aspect of NMR spectroscopy is spin-spin coupling.
Non-equivalent protons on adjacent carbon atoms influence the energy required to ‘flip’ the magnetic fields of each other. Let’s consider a simple molecule with two, non-equivalent proton environments.
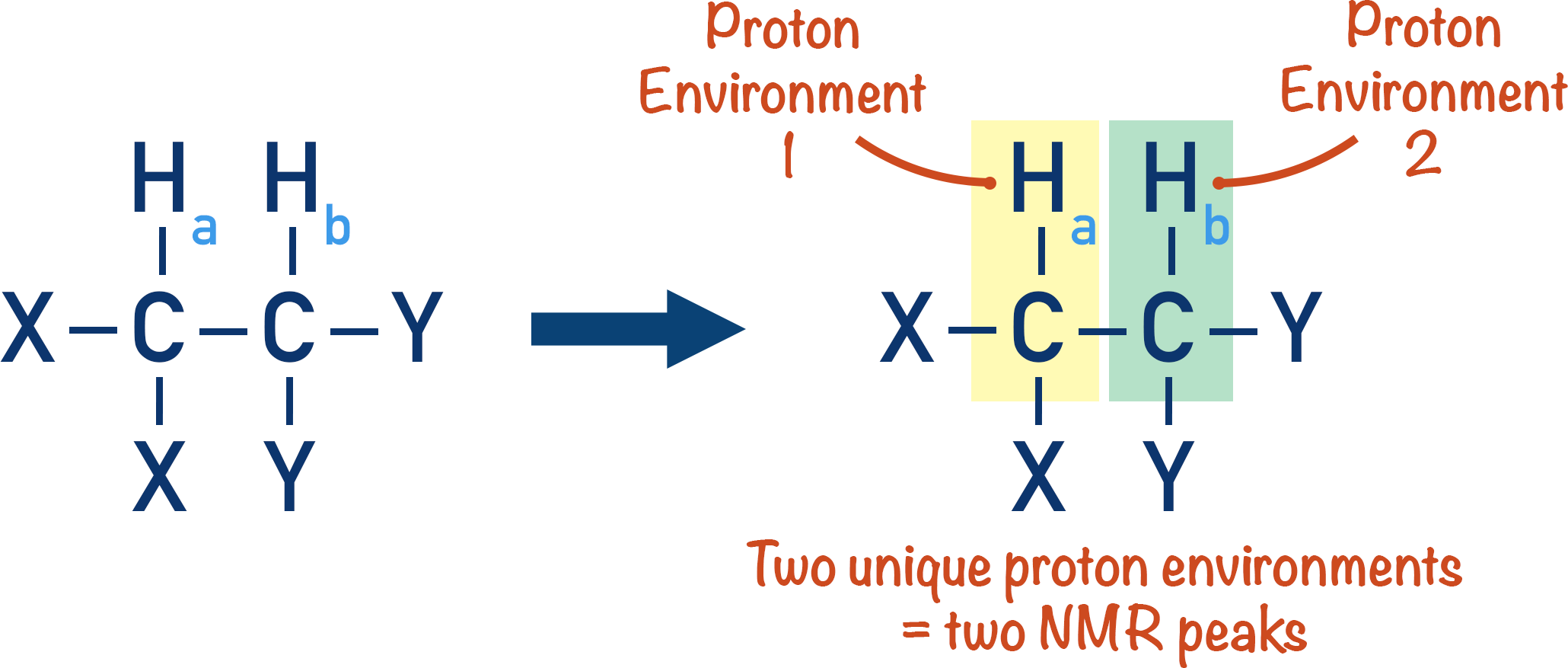
As non-equivalent protons have different environments, they will have different peaks in an NMR spectra.
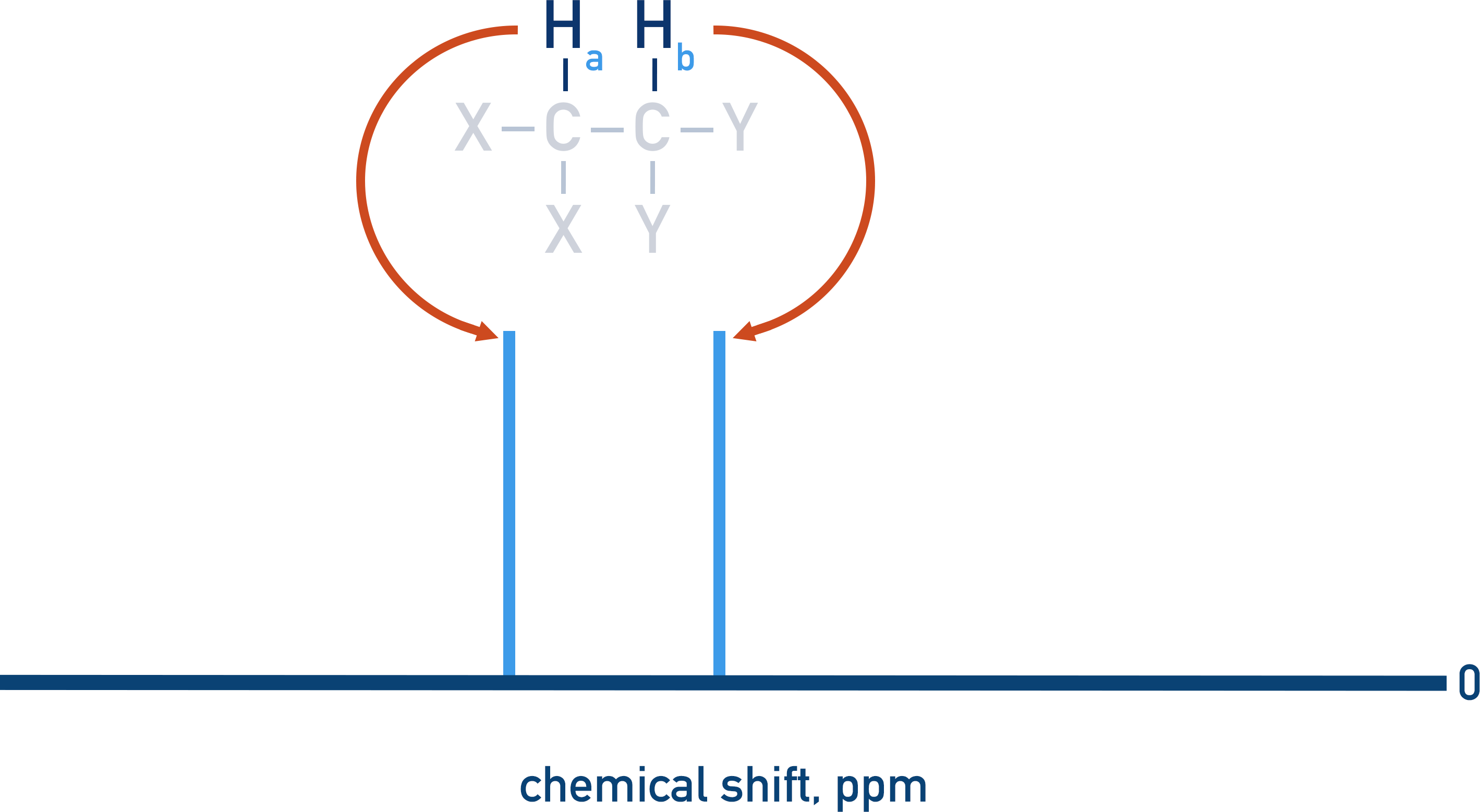
Each proton can either be aligned to the external magnetic field or non-aligned.
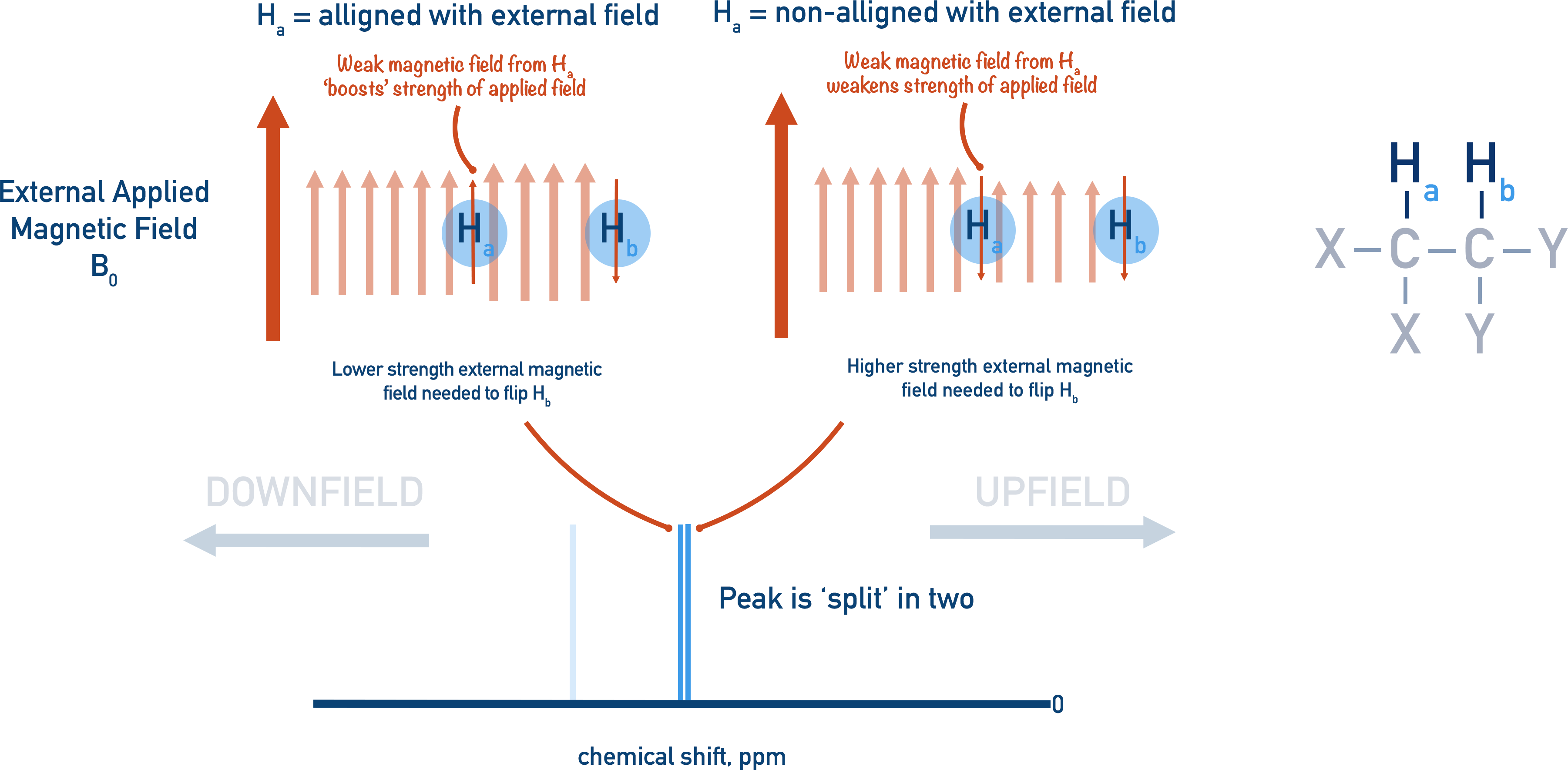
Let’s assume we are trying to ‘resonate’ Hb with an external magnetic field.
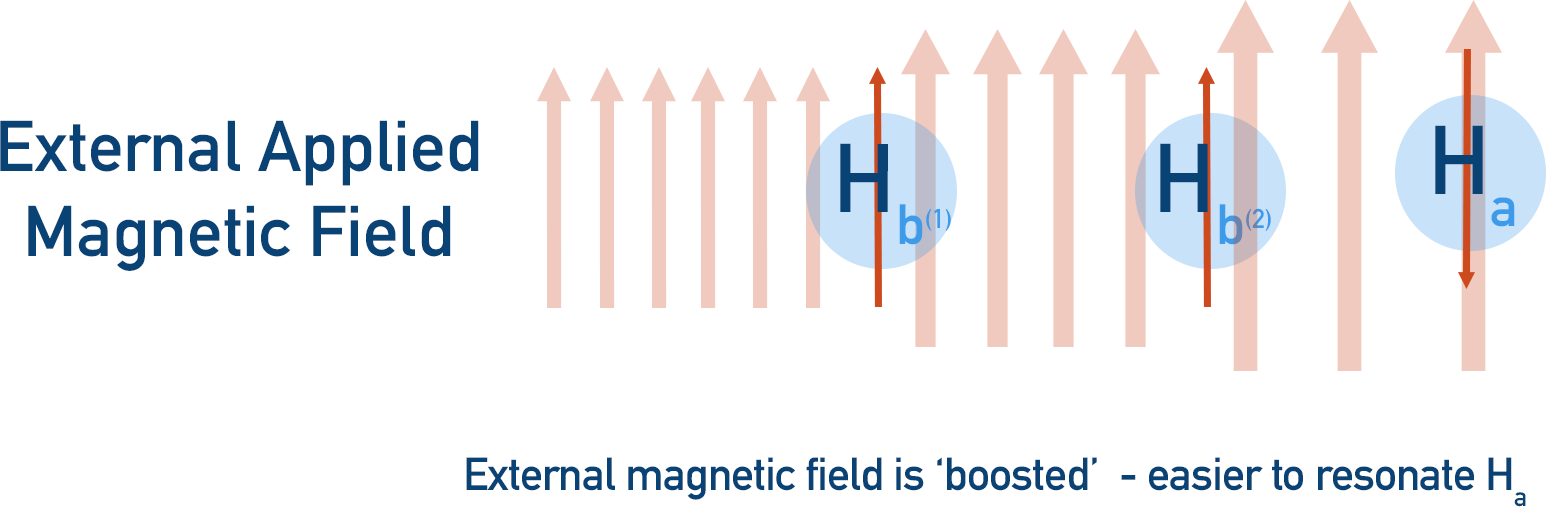
- If Ha is aligned to the external magnetic field, it contributes to the overall field being applied to Hb. So, less external magnetic field is required to resonate Hb, shifting Hb ’s further away from 0 (downfield).
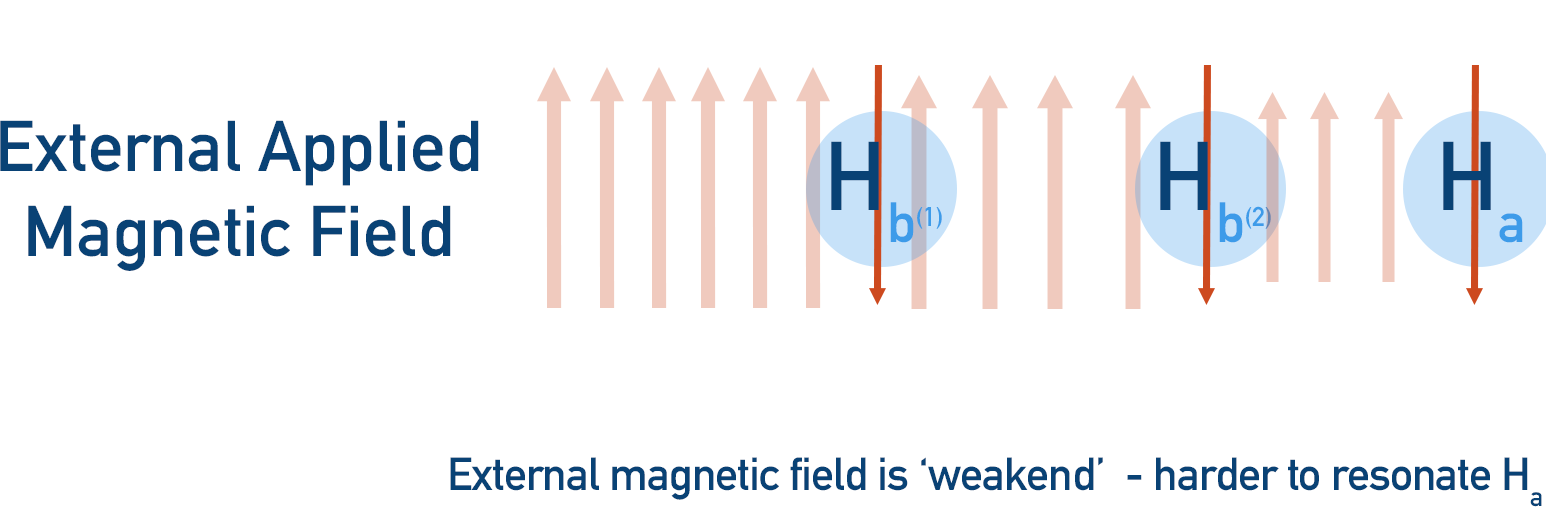
- If Ha is not aligned to the external magnetic field, it reduces the strength of the external magnetic field being applied to Hb. So, more external magnetic field is required to resonate Hb, shifting the Hb peak closer to 0 (upfield).
There is an equal chance of the magnetic field of Ha being aligned or non-aligned to the external magnetic field.
- 50% of the time Ha’s magnetic field will work with the external magnetic field, helping resonate Hb (shifting Hb’s peak downfield).
- 50% of the time Ha’s magnetic field will work against the external magnetic field, making it harder to resonate Hb (shifting Hb’s peak upfield).
- This is spin-spin coupling – the interaction of protons on adjacent, non-equivalent carbon atoms.
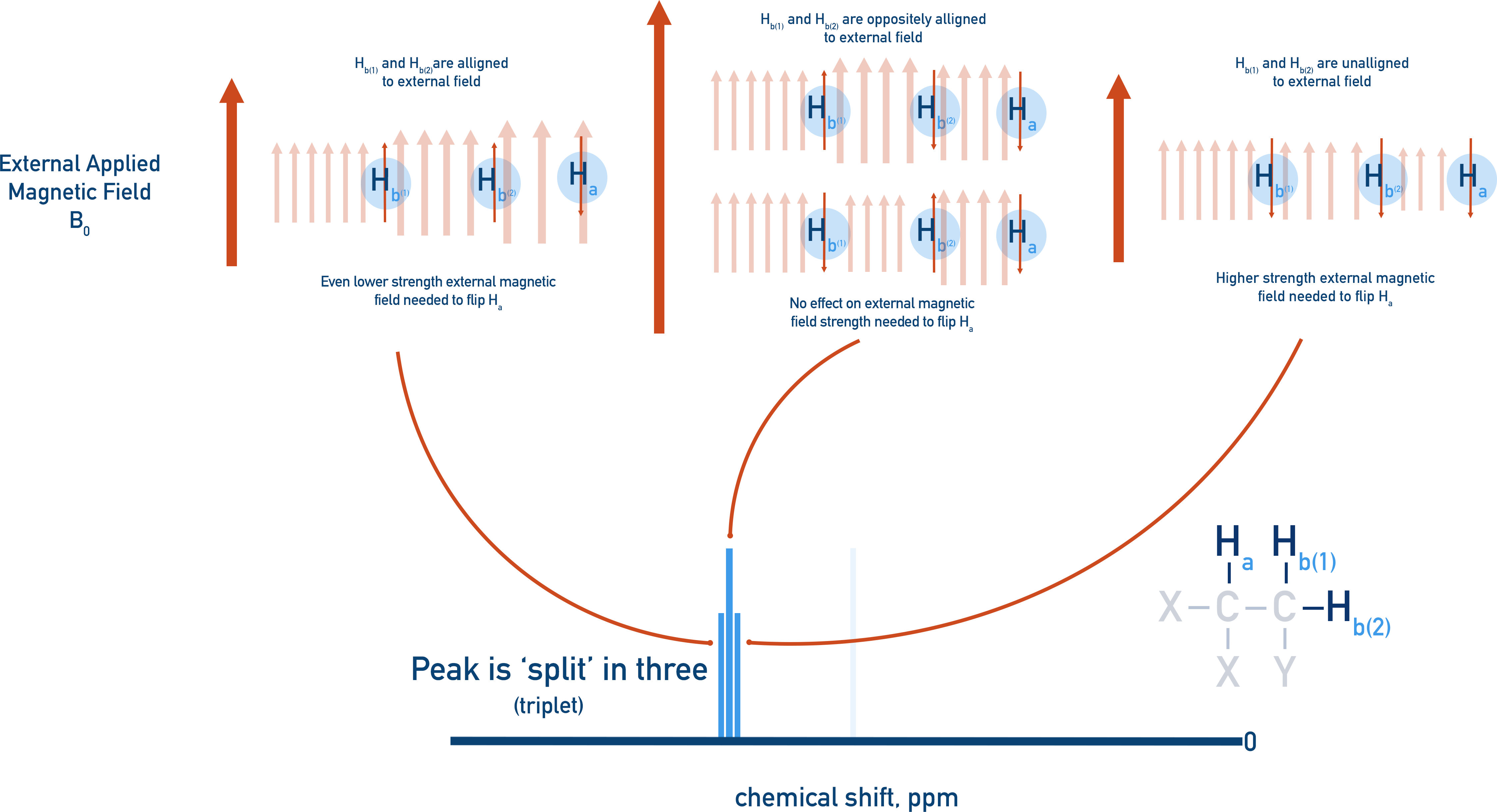
The Hb peak in the spectra is now ‘split’ into two lines: one line for when Ha’s magnetic field ‘helps’ resonate it and one line for when Ha’s magnetic field makes it harder to resonate it. This is referred to as a splitting pattern, and the unique splitting patterns that arise in a spectra can give useful insight into the bonding arrangements in the sample.
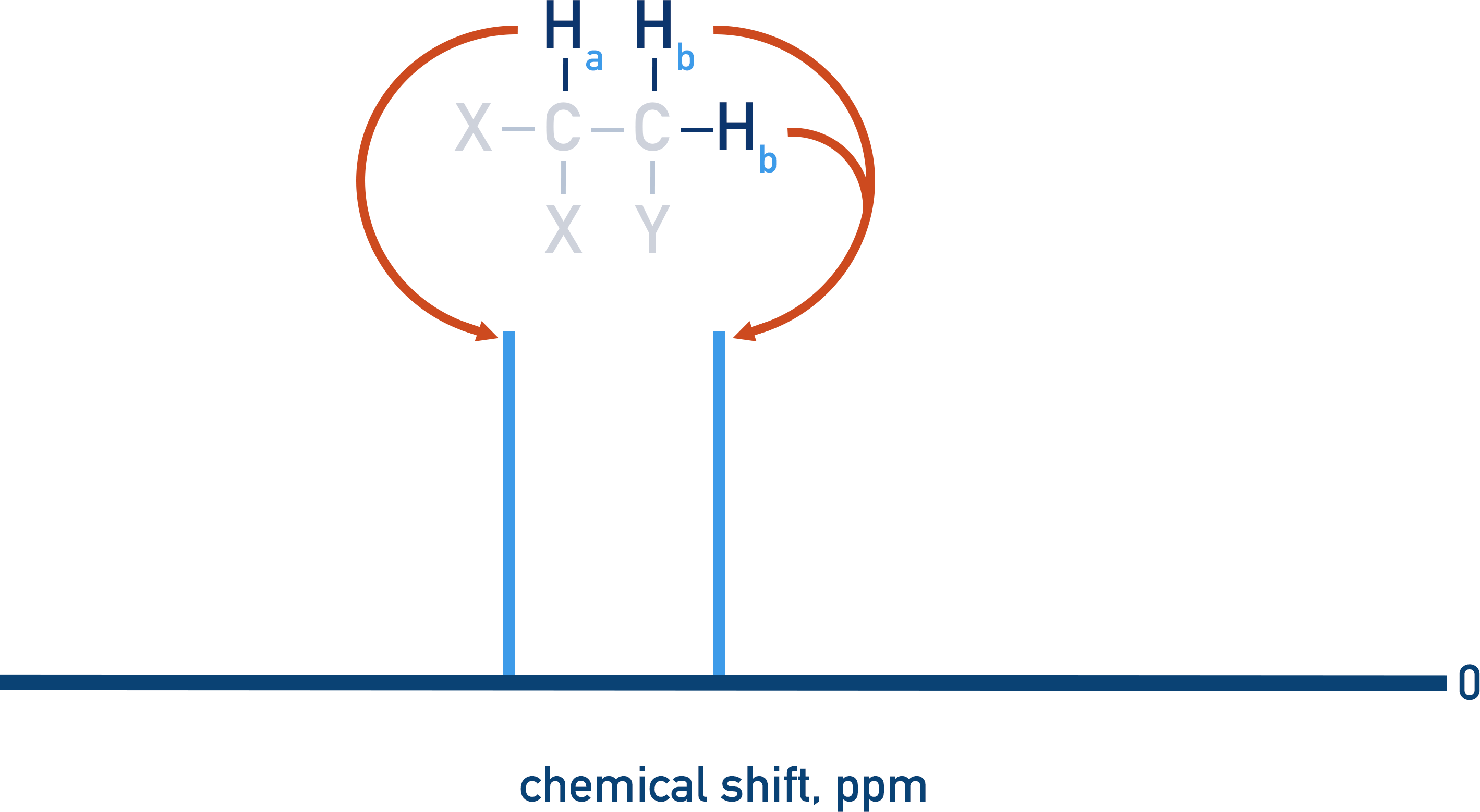
Equivalent protons are unable to spin-spin couple, meaning any split peaks on a spectra are caused by neighbouring protons on adjacent, non-equivalent carbon atoms.
The more protons there are in a neighbouring environment, the more ways a peak can be split. For example, if we take the same molecule as above but now put two protons in one of the enivronments, the splitting that occurs is more complex.
If we try and make Ha resonate, the two Hb protons can interfere with the applied magnetic field, causing Ha’s peak to be spilt into three (a triplet).
- Both Hb protons could be aligned with the external magnetic field, shifting Ha’s resonance downfield (further from 0).
- Both Hb protons could be unaligned with the magnetic field, shifting Ha’s resonance upfield (closer to 0).
- The two Hb protons could have opposite magnetic fields, effectively cancelling each other out. In this case, the resonance of Ha remains unshifted – it’s only affected by the external magnetic field.
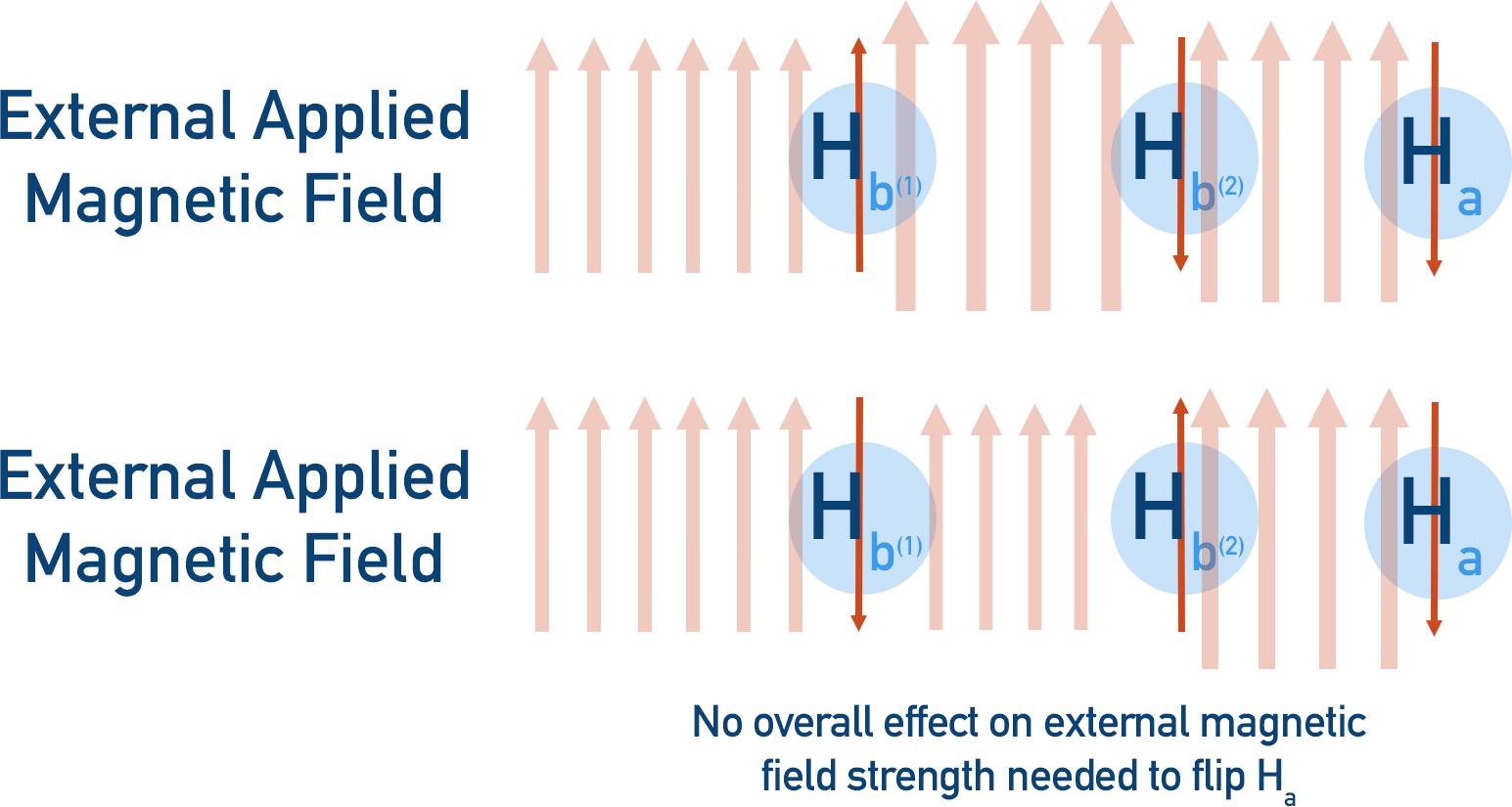
It’s more likely that the two Hb protons will have opposite spins rather than the same spin, meaning the intensity of the three splits are different.
n+1 rule
The number of times a peak can be split is dependent on the number of protons bonded to adjacent, non-equivalent carbon atoms.
Where n is the number of protons bonded to adjacent, non-equivalent carbon atoms, n+1 is the number of times a peak will be split.
This is useful to know when analysing spectra as it means the number of hydrogen atoms bonded to adjacent, non-equivalent carbon atoms can be determined from peak splitting.
Splitting pattern
1 (singlet) n +1 = 1, n must be 0
2 (doublet) n + 1 = 2, n must be 1
3 (triplet) n + 1 = 3, n must be 2
4 (quartet) n + 1 = 4, n must be 3
5 (quintet) n + 1 = 5, n must be 4
We’ve launched our new site! 🎉
Course-specific notes with built-in search!
AP • A-Level (AQA • CIE • Edexcel • OCR) • IB • NCERT 11 + 12
over 750+ new pages and 3,500 images.
Visit the new homepage